Chapter 6: Short-lived Climate Forcers
This chapter should be cited as:
Szopa, S., V. Naik, B. Adhikary, P. Artaxo, T. Berntsen, W.D. Collins, S. Fuzzi, L. Gallardo, A. Kiendler-Scharr, Z. Klimont, H. Liao, N. Unger, and P. Zanis, 2021: Short-Lived Climate Forcers. In Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change [Masson-Delmotte, V., P. Zhai, A. Pirani, S.L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M.I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J.B.R. Matthews, T.K. Maycock, T. Waterfield, O. Yelekçi, R. Yu, and B. Zhou (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 817–922, doi: 10.1017/9781009157896.008.
Executive Summary
Short-lived climate forcers (SLCFs) affect climate and are, in most cases, also air pollutants. They include aerosols (sulphate, nitrate, ammonium, carbonaceous aerosols, mineral dust and sea spray), which are also called particulate matter (PM), and chemically reactive gases (methane, ozone, some halogenated compounds, nitrogen oxides, carbon monoxide, non-methane volatile organic compounds, sulphur dioxide and ammonia). Except for methane and some halogenated compounds whose lifetimes are about a decade or more, SLCF abundances are spatially highly heterogeneous since they only persist in the atmosphere from a few hours to a couple of months. SLCFs are either radiatively active or influence the abundances of radiatively active compounds through chemistry (chemical adjustments), and their climate effect occurs predominantly in the first two decades after their emission or formation. They can have either a cooling or warming effect on climate, and they also affect precipitation and other climate variables. Methane and some halogenated compounds are included in climate treaties, unlike the other SLCFs that are nevertheless indirectly affected by climate change mitigation since many of them are often co-emitted with CO2 in combustion processes. This chapter assesses the changes, in the past and in a selection of possible futures, of the emissions and abundances of individual SLCFs primarily on global to continental scales, and how these changes affect the Earth’s energy balance through radiative forcing and feedback in the climate system. The attribution of climate and air-quality changes to emissions sectors and regions, and the effects of SLCF mitigations defined for various environmental purposes, are also assessed.
Recent Evolution in Short-lived Climate Forcer (SLCF) Emissions and Abundances
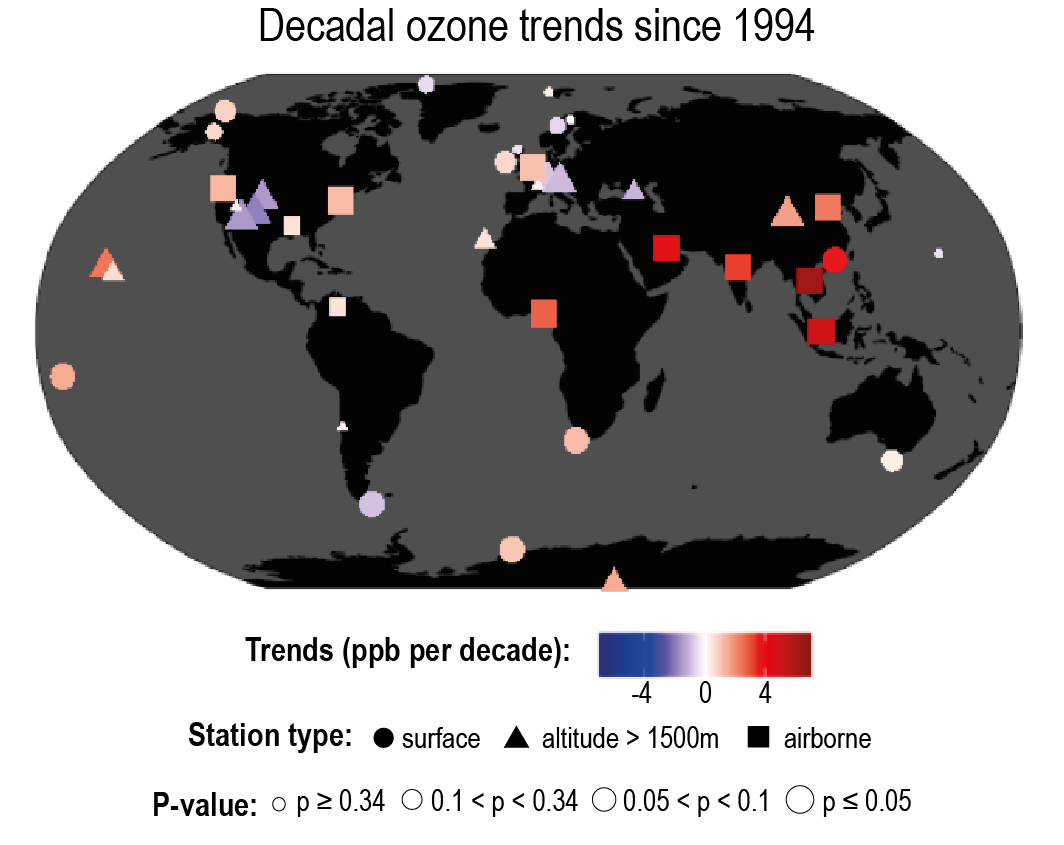
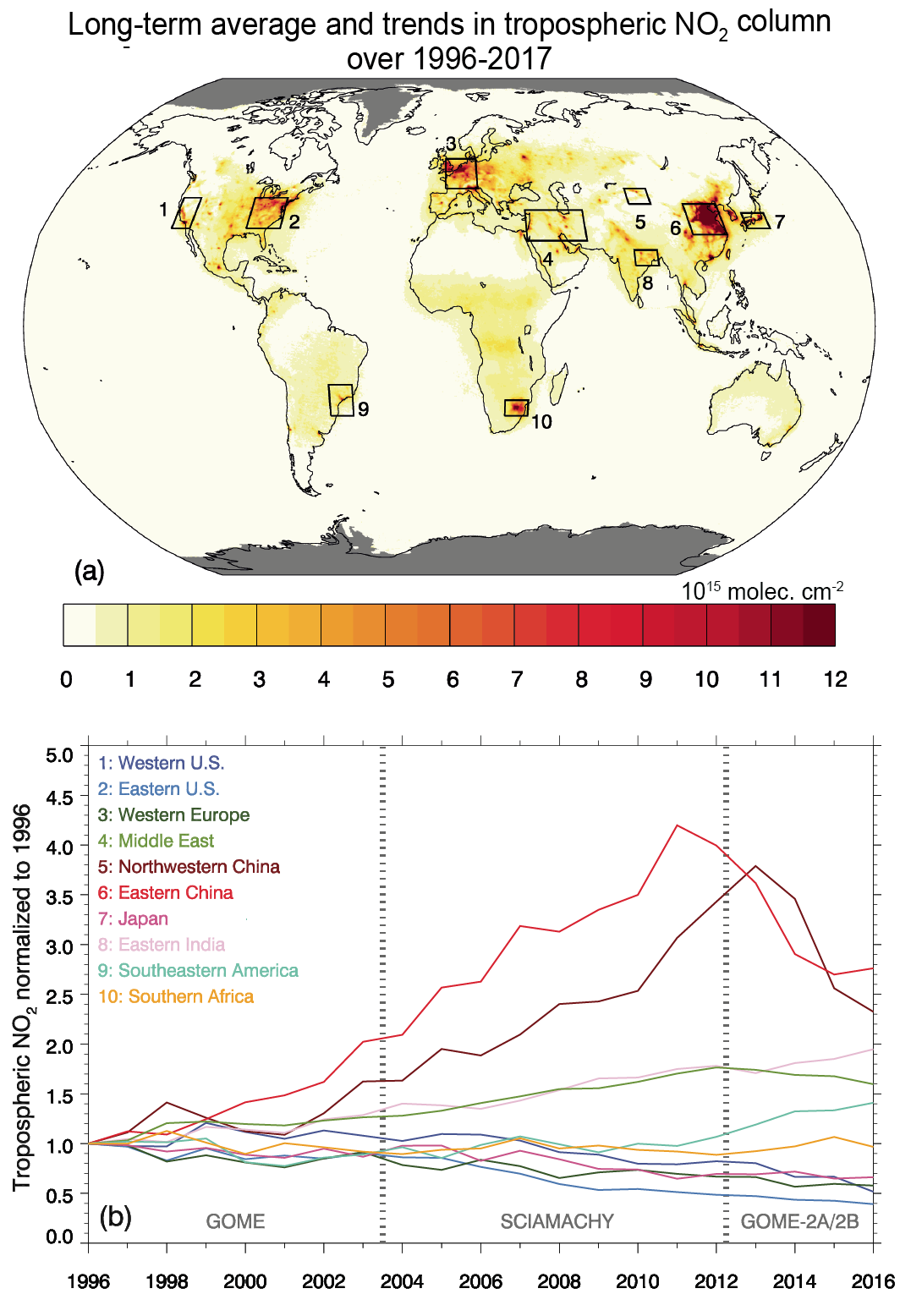
Over the last decade (2010–2019), strong shifts in the geographical distribution of emissions have led to changes in atmospheric abundances of highly variable SLCFs (high confidence). Evidence from satellite and surface observations shows strong regional variations in trends of ozone (O3), aerosols and their precursors (high confidence). In particular, tropospheric columns of nitrogen dioxide (NO2) and sulphur dioxide (SO2) continued to decline over North America and Europe (high confidence), and to increase over Southern Asia (medium confidence), but have declined over Eastern Asia (high confidence). Global carbon monoxide (CO) abundance has continued to decline (high confidence). The concentrations of hydrofluorocarbons (HFCs) are increasing (high confidence). Global carbonaceous aerosol budgets and trends remain poorly characterized due to limited observations, but sites representative of background conditions have reported multi-year declines in black carbon (BC) over several regions of the Northern Hemisphere. {6.2, 6.3, 2.2.4, 2.2.5, 2.2.6}
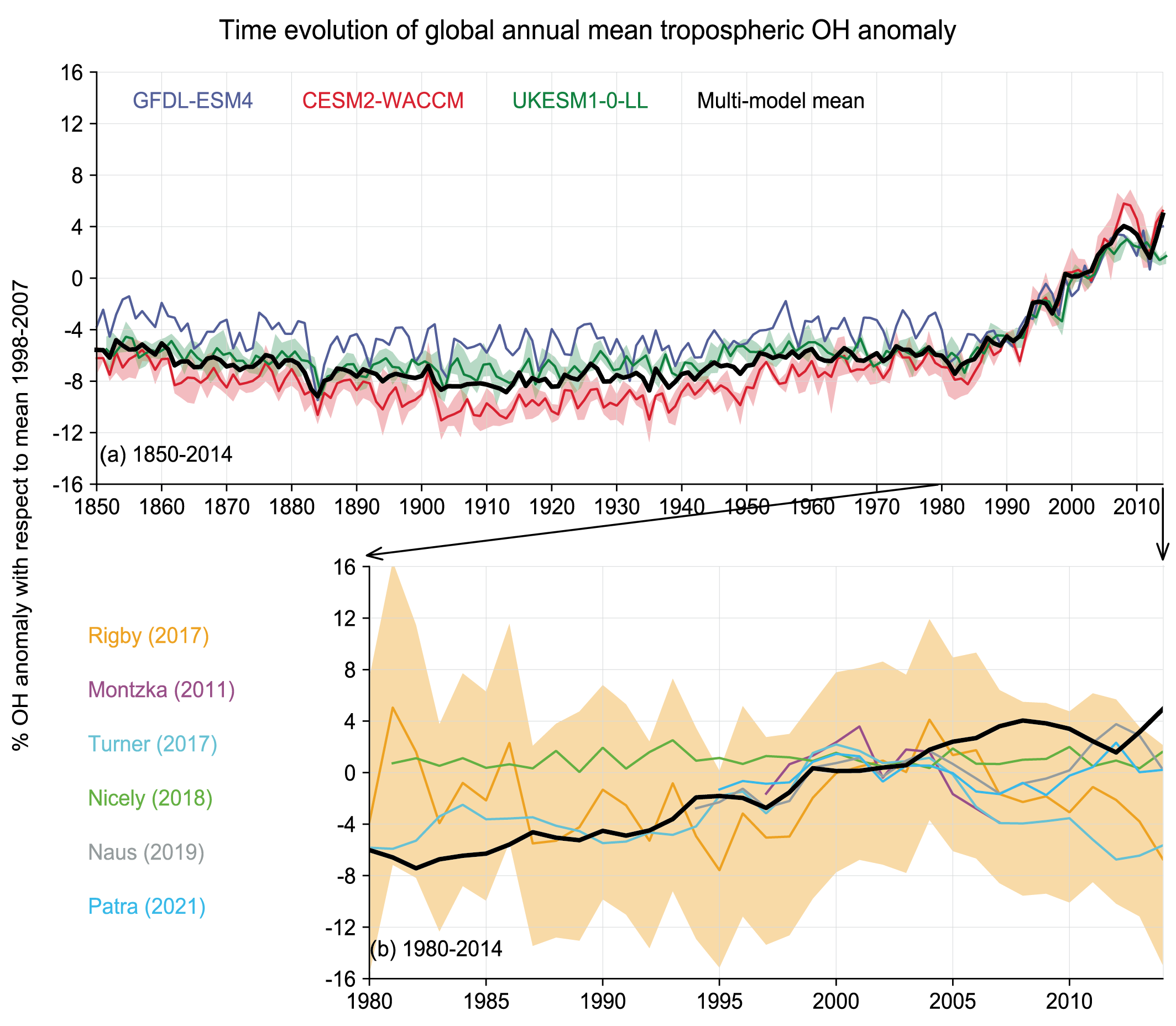
There is no significant trend in the global mean tropospheric concentration of hydroxyl (OH) radical – the main sink for many SLCFs, including methane (CH4) – from 1850 up to around 1980 (low confidence) but OH has remained stable or exhibited a positive trend since the 1980s (medium confidence). Global OH cannot be measured directly and is inferred from Earth system and chemistry–climate models (ESMs, CCMs) constrained by emissions and from observationally constrained inversion methods. There is conflicting information from these methods for the 1980–2014 period. ESMs and CCMs concur on a positive trend since 1980 (about a 9% increase over 1980–2014) and there is medium confidence that this trend is mainly driven by increases in global anthropogenic (human-caused) nitrogen oxide (NOx) emissions and decreases in anthropogenic CO emissions. The observation-constrained methods suggest either positive trends or the absence of trends based on limited evidence and medium agreement. Future changes in global OH, in response to SLCF emissions and climate change, will depend on the interplay between multiple offsetting drivers of OH. {6.3.6 and Cross-Chapter Box 5.1}
Effect of SLCFs on Climate and Biogeochemical Cycles
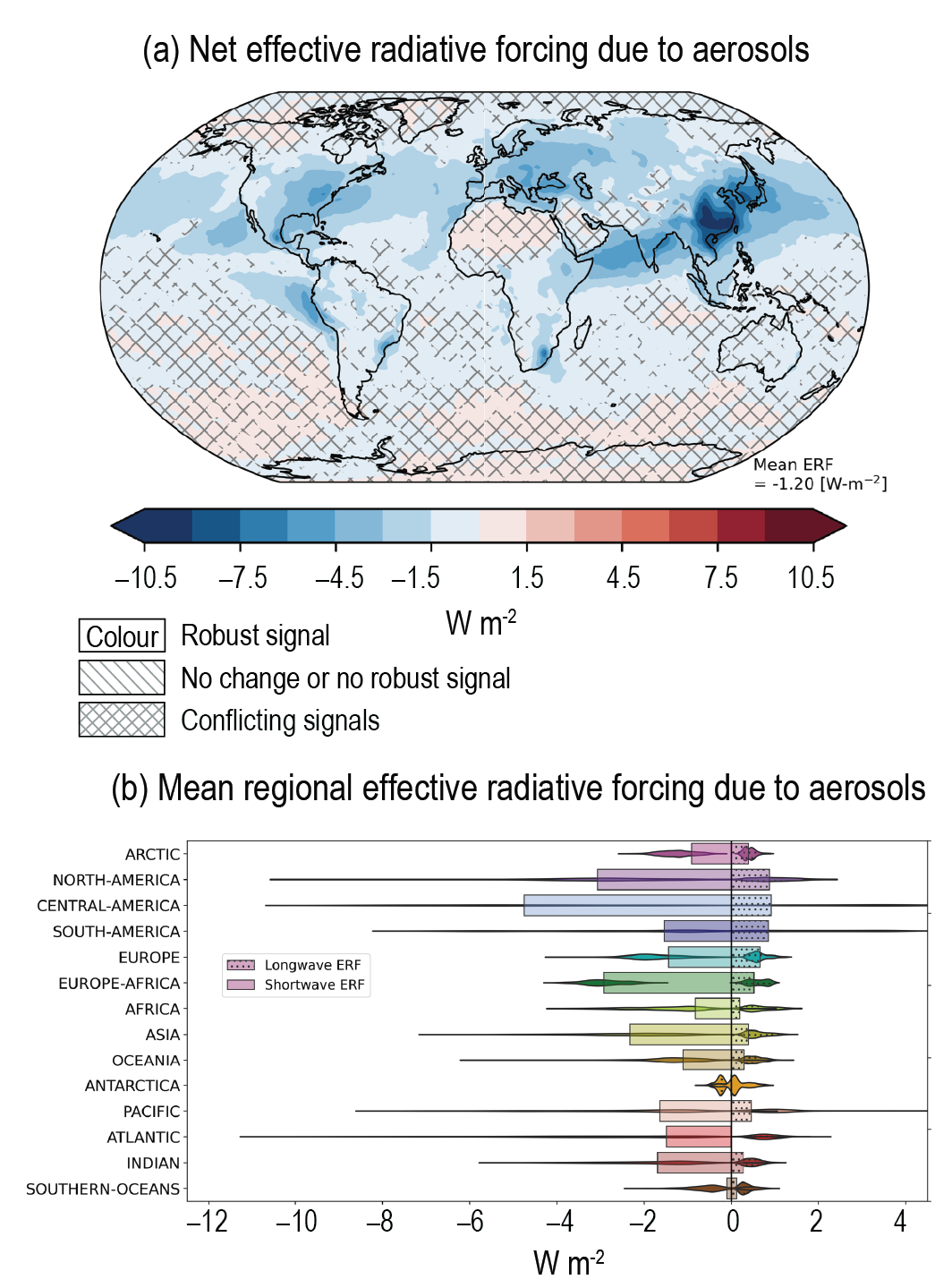
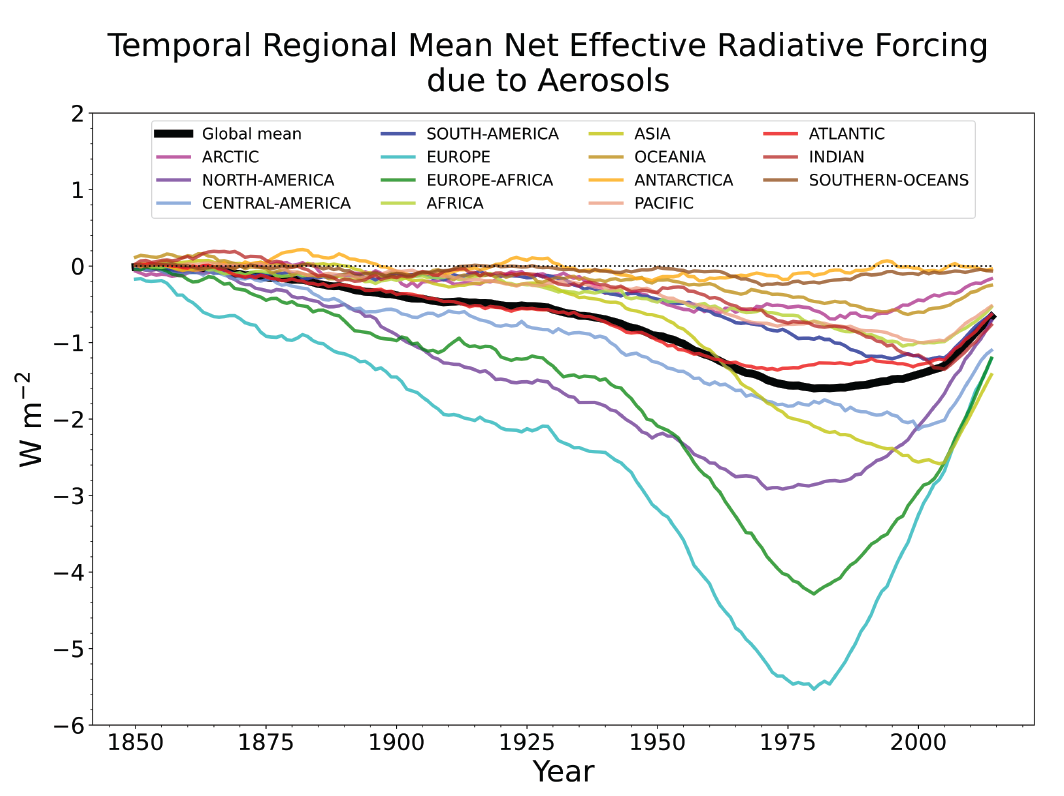
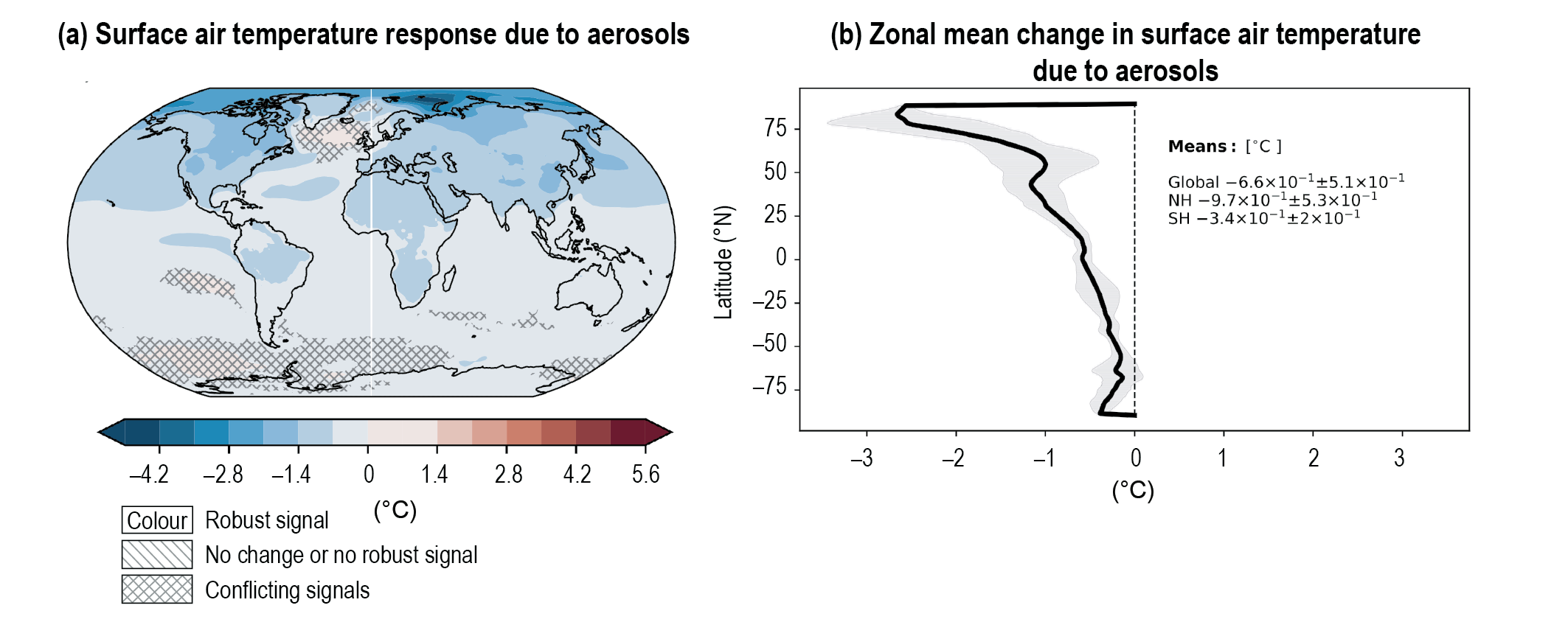
Over the historical period, changes in aerosols and their effective radiative forcing (ERF) have primarily contributed to a surface cooling, partly masking the greenhouse gas-driven warming (high confidence). Radiative forcings induced by aerosol changes lead to both local and remote temperature responses (high confidence). The temperature response preserves the south to north gradient of the aerosol ERF – hemispherical asymmetry – but is more uniform with latitude and is strongly amplified towards the Arctic (medium confidence). {6.4.1, 6.4.3}
Since the mid-1970s, trends in aerosols and their precursor emissions have led to a shift from an increase to a decrease of the magnitude of the negative globally averaged net aerosol ERF (high confidence). However, the timing of this shift varies by continental-scale region and has not occured for some finer regional scales. The spatial and temporal distribution of the net aerosol ERF from 1850 to 2014 is highly heterogeneous, with stronger magnitudes in the Northern Hemisphere (high confidence). {6.4.1}
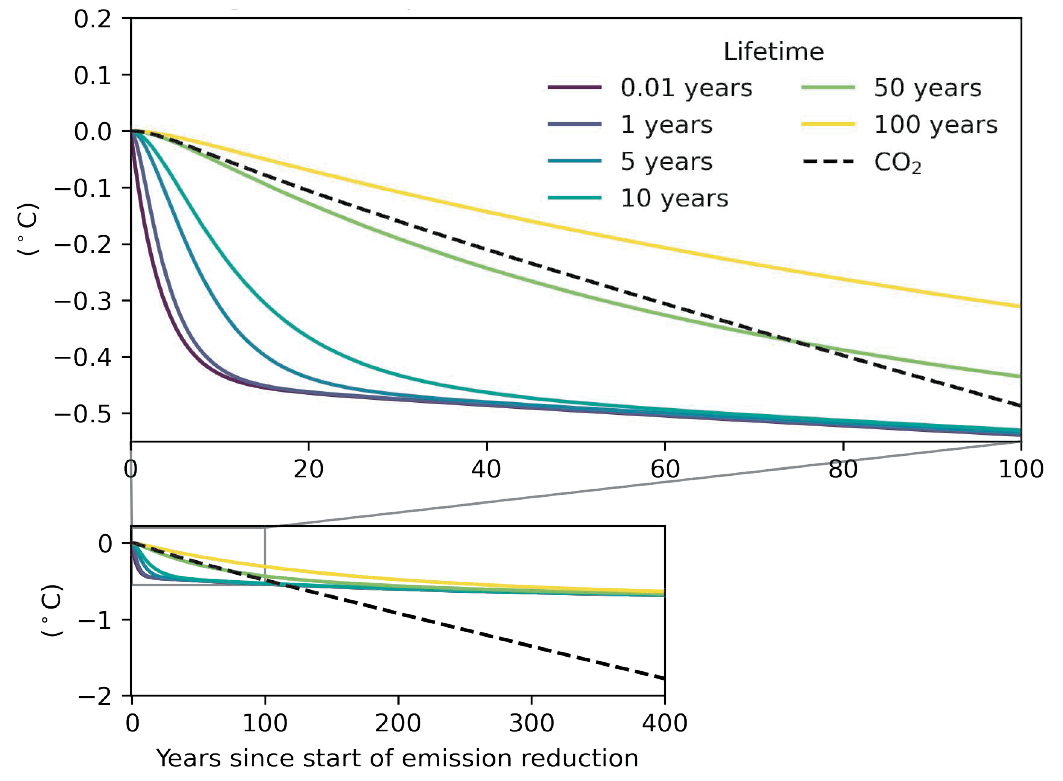
For forcers with short lifetimes (e.g., months) and not considering chemical adjustments, the response in surface temperature occurs strongly as soon as a sustained change in emissions is implemented, and that response continues to grow for a few years, primarily due to thermal inertia in the climate system (high confidence). Near its maximum, the response slows down but will then take centuries to reach equilibrium (high confidence). For SLCFs with longer lifetimes (e.g., a decade), a delay equivalent to their lifetimes is appended to the delay due to thermal inertia (high confidence). {6.6.1}
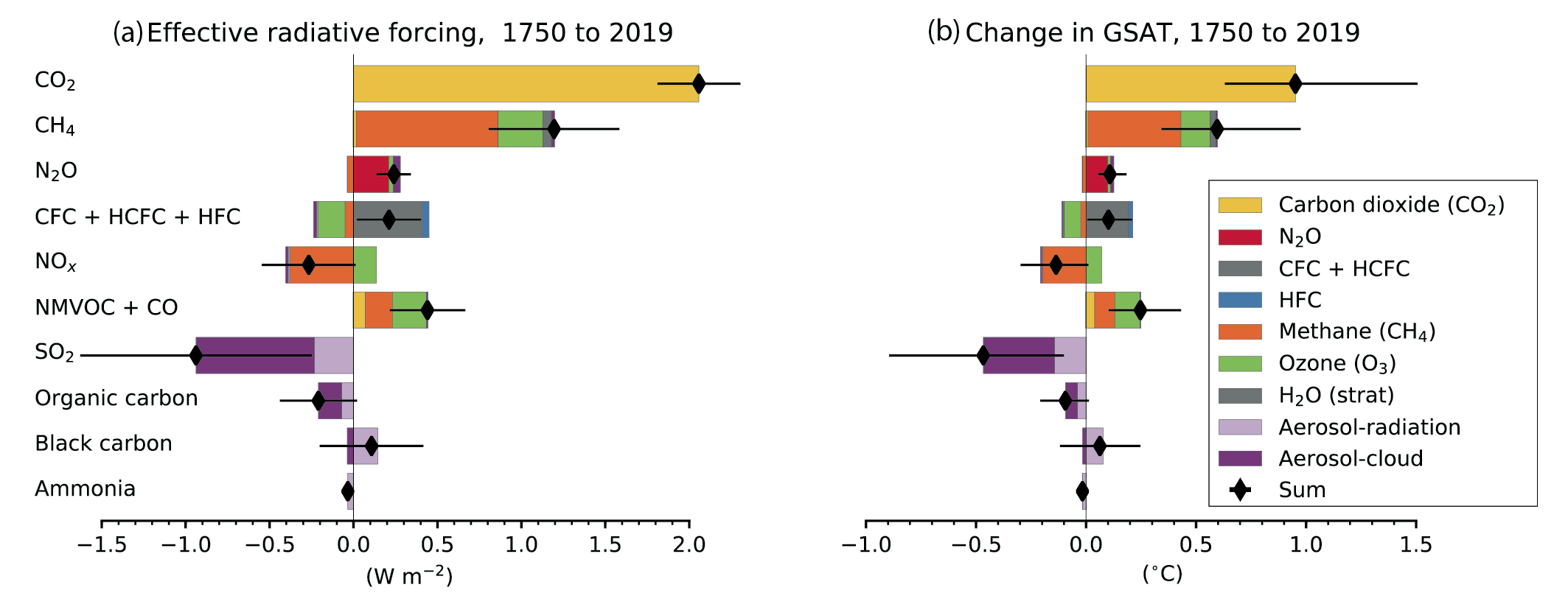
Over the 1750–2019 period, changes in SLCF emissions, especially of methane, NOxand SO2, have substantial effects on effective radiative forcing (ERF) (high confidence). The net global emissions-based ERF of NOx is negative and that of non-methane volatile organic compounds (NMVOCs) is positive, in agreement with the AR5 Assessment (high confidence). For methane, the emissions-based ERF is twice as high as the abundance-based ERF (high confidence) attributed to chemical adjustment mainly via ozone production. SO2 emissions changes make the dominant contribution to the ERF from aerosol–cloud interactions (high confidence). Over the 1750–2019 period, the contributions from the emitted compounds to changes in global surface air temperature (GSAT) broadly match their contributions to the ERF (high confidence). Since a peak in emissions-induced SO2 eRF has already occurred recently and since there is a delay in the full GSAT response, changes in SO2 emissions have a slightly larger contribution to GSAT change than CO2 emissions, relative to their respective contributions to ERF. {6.4.2, 6.6.1 and 7.3.5}
Reactive nitrogen, ozone and aerosols affect terrestrial vegetation and the carbon cycle through deposition and effects on large-scale radiation (high confidence). However, the magnitude of these effects on the land carbon sink, ecosystem productivity and hence their indirect CO2 forcing remain uncertain due to the difficulty in disentangling the complex interactions between the individual effects. As such, these effects are assessed to be of second order in comparison to the direct CO2 forcing (high confidence), but effects of ozone on terrestrial vegetation could add a substantial (positive) forcing compared with the direct ozone forcing (low confidence). {6.4.4}
Climate feedbacks induced from changes in emissions, abundances or lifetimes of SLCFs mediated by natural processes or atmospheric chemistry are assessed to have an overall cooling effect (low confidence), that is, a total negative feedback parameter of –0.20 [–0.41 to +0.01] W m–2°C−1. These non-CO2 biogeochemical feedbacks are estimated from ESMs, which have advanced since AR5 to include a consistent representation of biogeochemical cycles and atmospheric chemistry. However, process-level understanding of many chemical and biogeochemical feedbacks involving SLCFs, particularly natural emissions, is still emerging, resulting in low confidence in the magnitude andsign of most SLCF climatefeedback parameters. {6.2.2, 6.4.5}
Future Projections for Air Quality Considering Shared Socio-economic Pathways (SSPs)
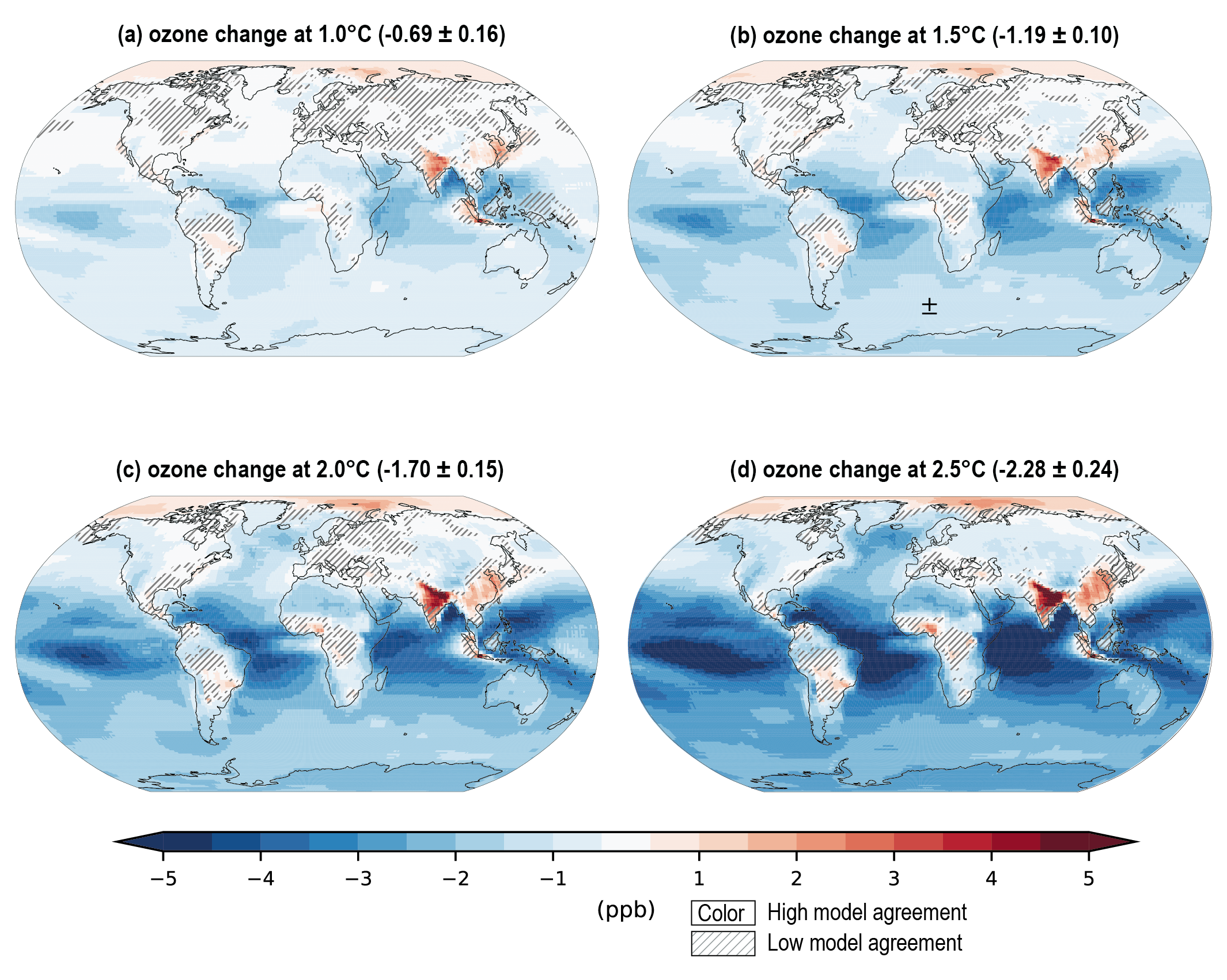
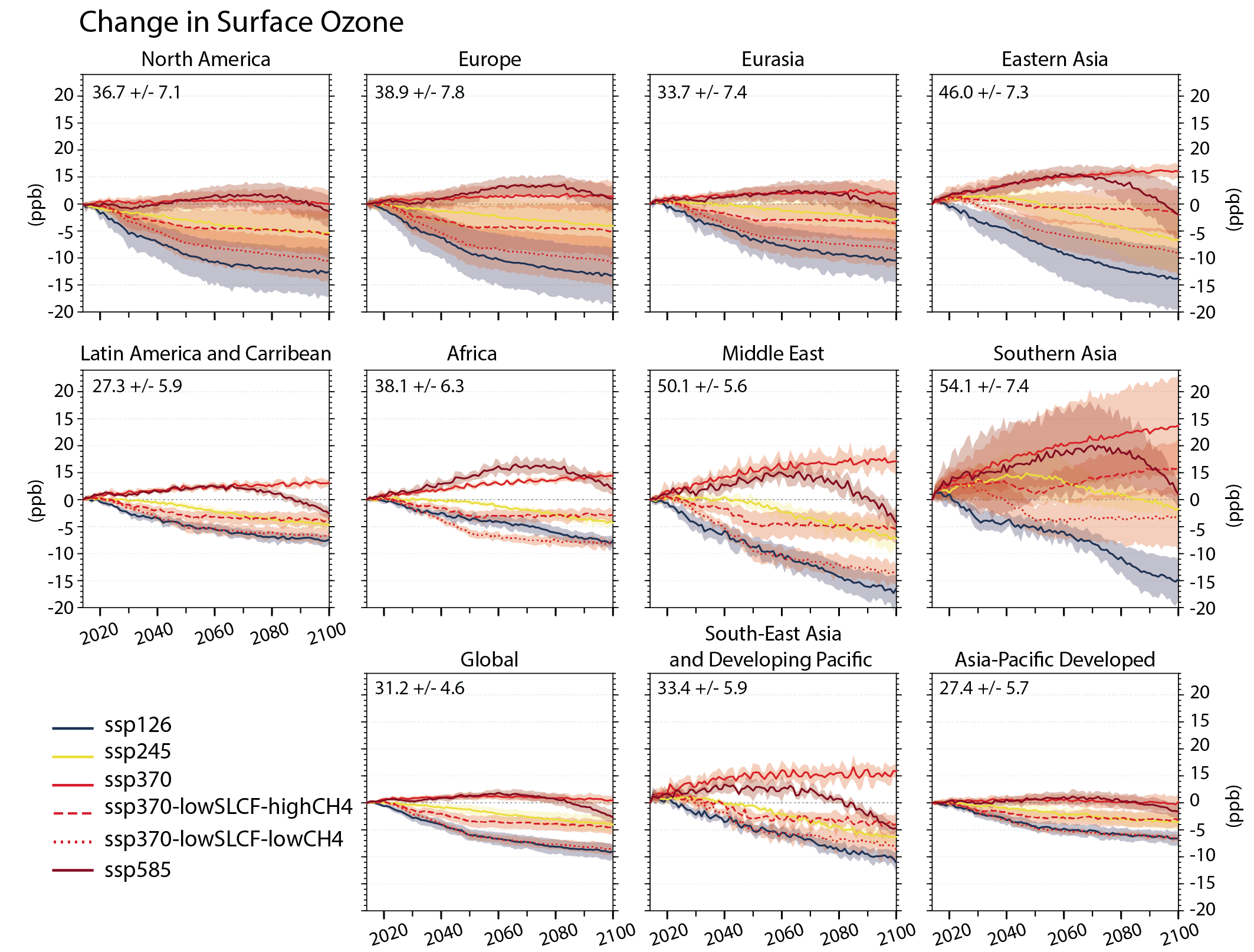
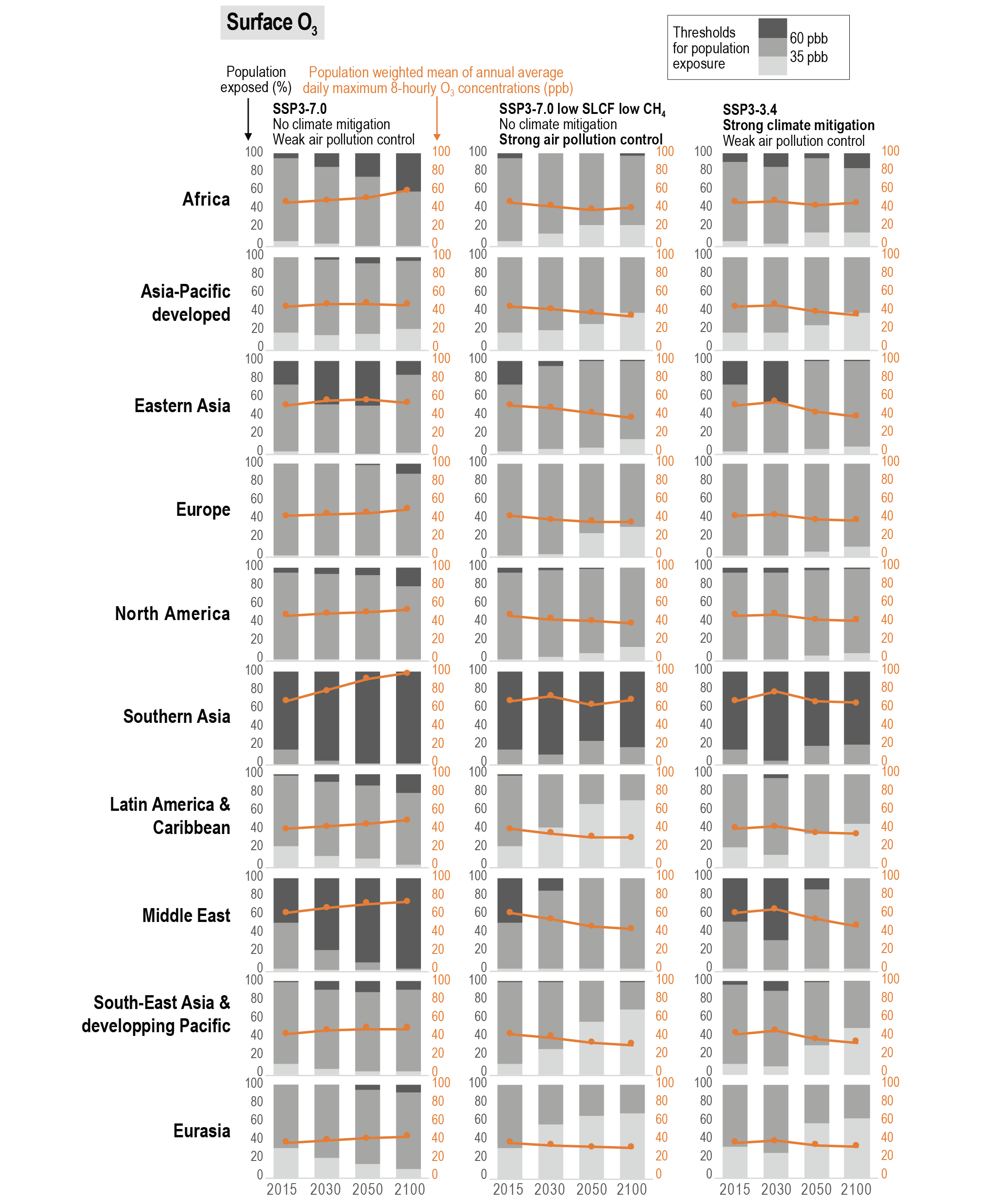
Future air quality (in term of surface ozone and PM concentrations) on global to local scales will be primarily driven by changes in precursor emissions as opposed to climate change (high confidence) and climate change is projected to have mixed effects. A warmer climate is expected to reduce surface ozone in regions remote from pollution sources (high confidence) but is expected to increase it by a few parts per billion over polluted regions, depending on ozone precursor levels (medium tohigh confidence). Future climate change is expected to have mixed effects, positive or negative, with an overall low effect, on global surface PM and more generally on the aerosol global burden (medium confidence) , but stronger effects are not excluded in regions prone to specific meteorological conditions (low confidence). Overall, there islow confidence in the response of surface ozone and PM to future climate change due to the uncertainty in the response of the natural processes (e.g., stratosphere–troposphere exchange, natural precursor emissions, particularly including biogenic volatile organic compounds, wildfire-emitted precursors, land and marine aerosols, and lightning NOx) to climate change. {6.3, 6.5}
The SSPs span a wider range of SLCF emissions than the Representative Concentration Pathways (RCPs), representing better the diversity of future options in air pollution management (high confidence). In the SSPs, the socio-economic assumptions and climate change mitigation levels primarily drive future emissions, but the SLCF emissions trajectories are also steered by varying levels of air pollution control originating from the SSP narratives, independently from climate change mitigation. Consequently, SSPs consider a large variety of regional ambitions and effectiveness in implementing air pollution legislation and result in wider range of future air pollution levels and SLCF-induced climate effects. {6.7.1}
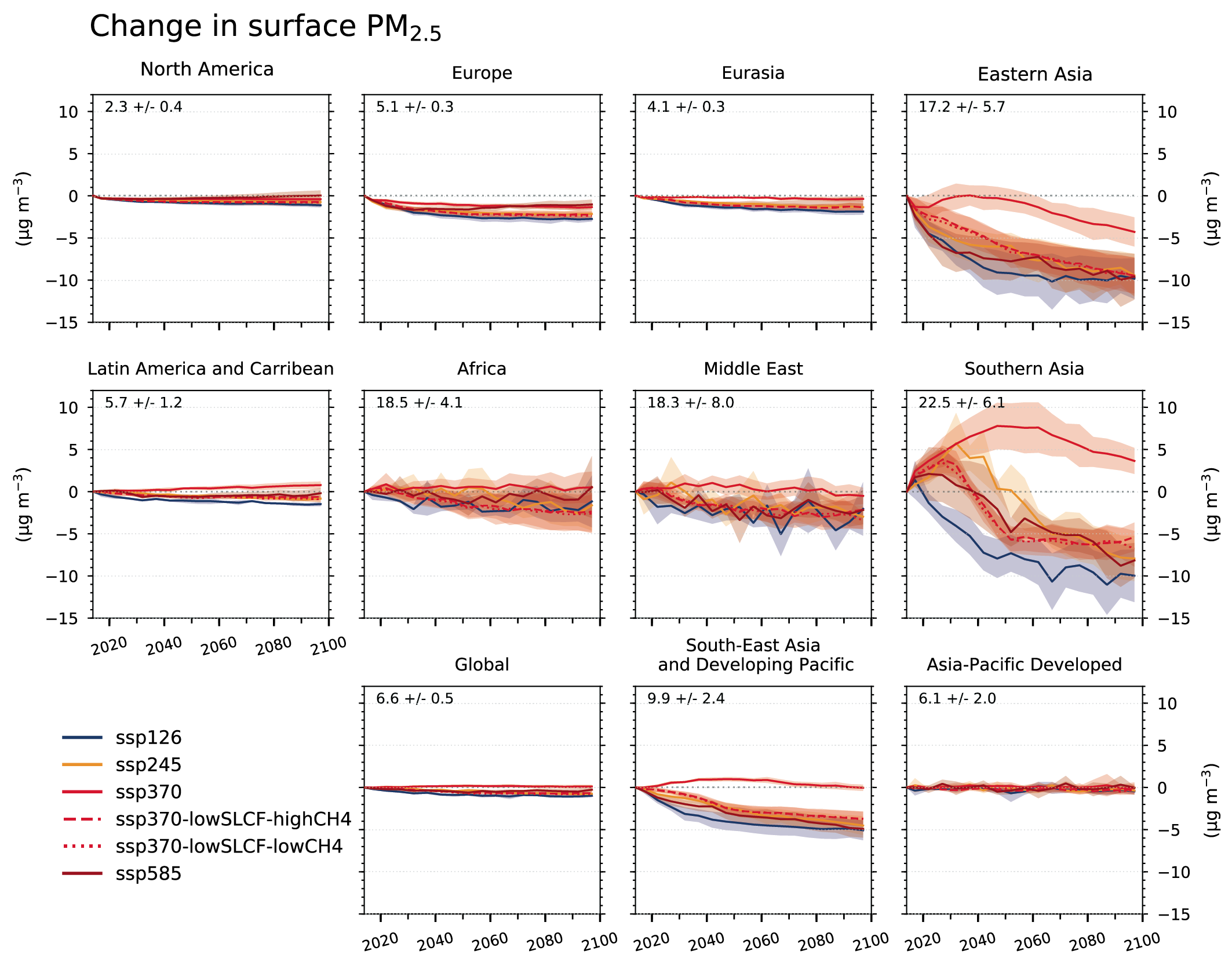
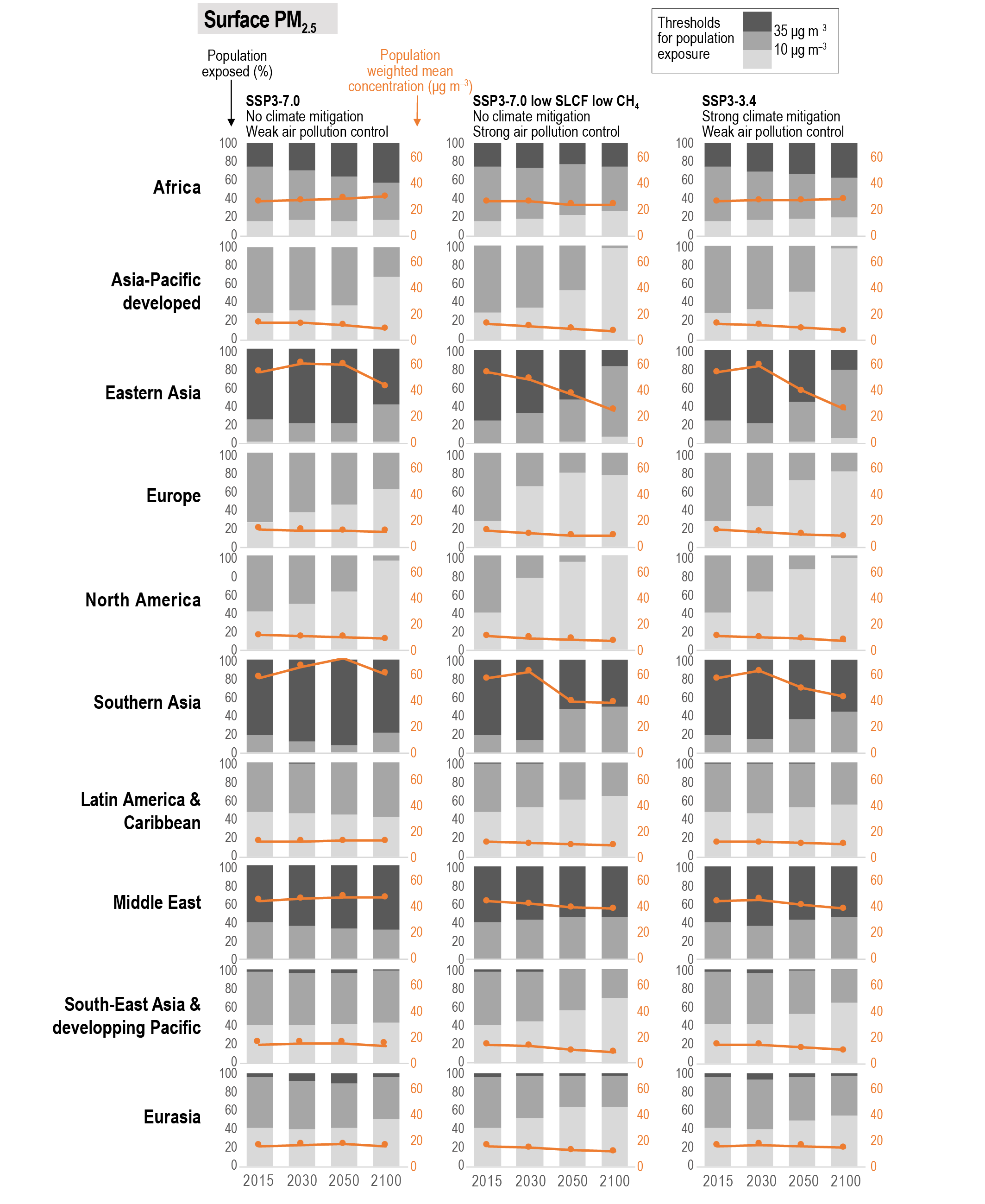
Air pollution projections range from strong reductions in global surface ozone and PM (e.g., SSP1-2.6, with strong mitigation of both air pollution and climate change) to no improvement and even degradation (e.g., SSP3-7.0 without climate change mitigation and with only weak air pollution control) (high confidence). Under the SSP3-7.0scenario, PM levels are projected to increase until 2050 over large parts of Asia, and surface ozone pollution is projected to worsen over all continental areas through 2100 (high confidence). Without climate change mitigation but with stringent air pollution control (SSP5-8.5), PM levels decline through 2100, but high methane levels hamper the decline in global surface ozone at least until 2080 (high confidence). {6.7.1}
Future Projections of the Effect of SLCFs on GSAT in the Core SSPs
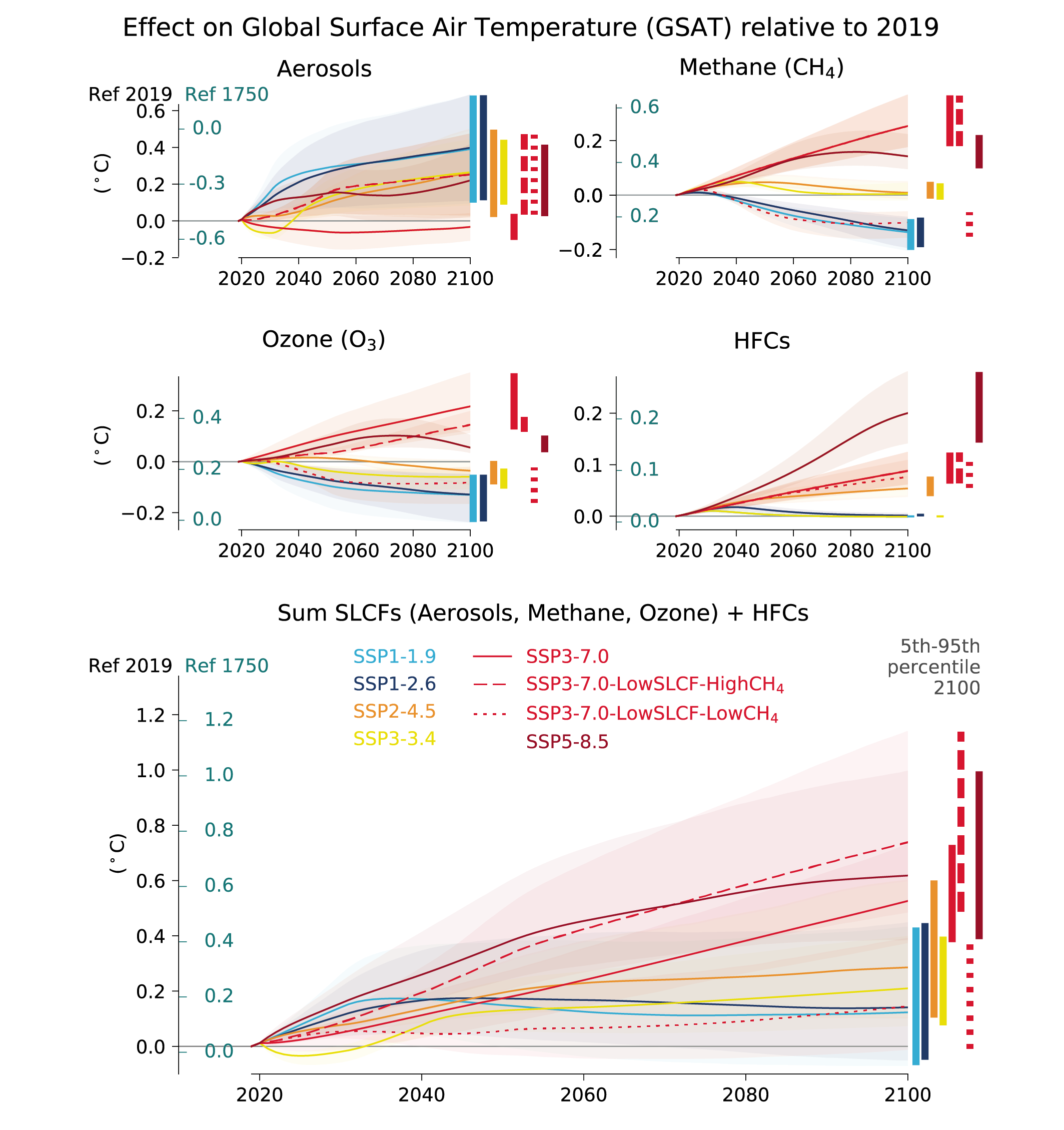
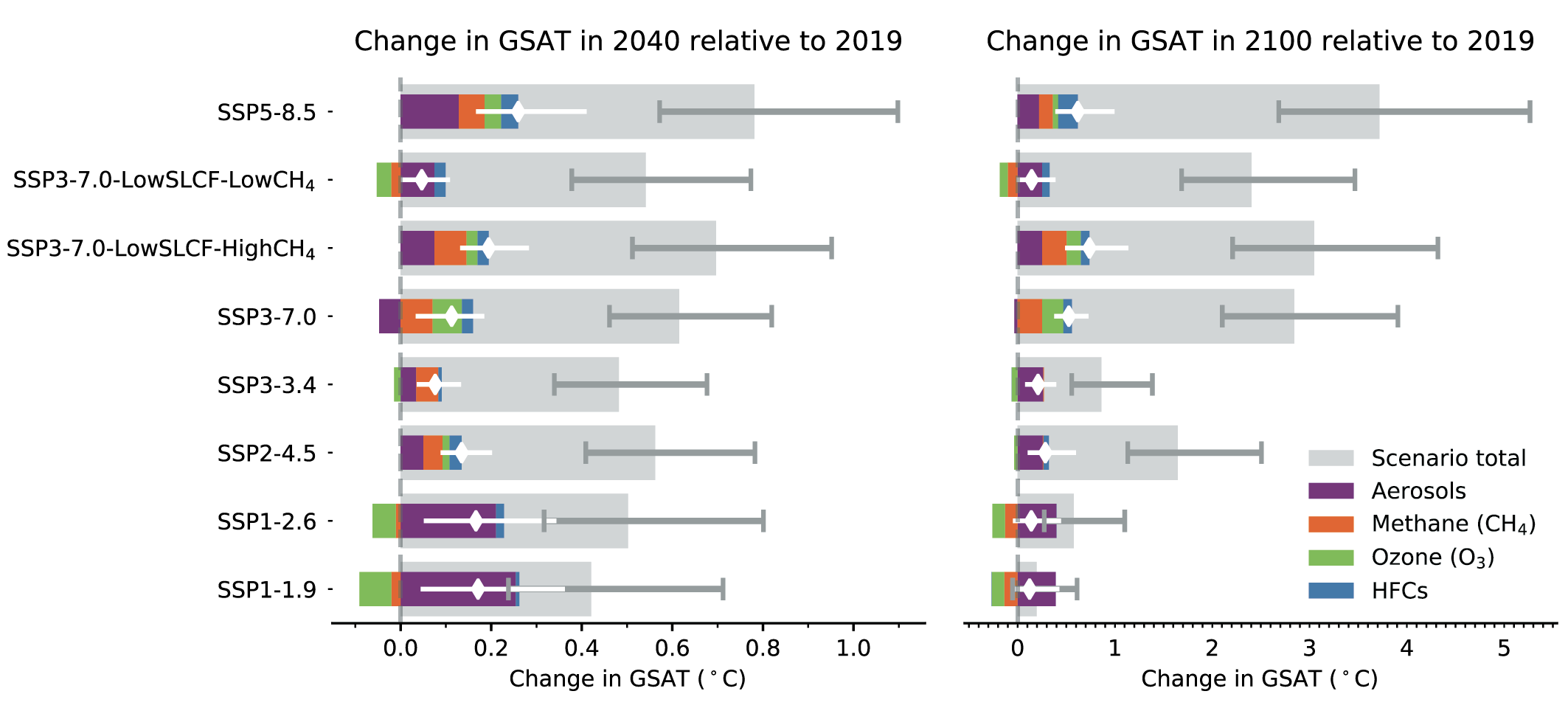
In the next two decades, it is very likely that the SLCF emissions changes in the WGI core set of SSPs will cause a warming relative to 2019, whatever the SSPs, in addition to the warming from long-lived greenhouse gases. The net effect of SLCF and hydrofluorocarbon (HFC) changes on GSAT across the SSPsis a likely warming of 0.06°C–0.35°C in 2040 relative to 2019. Warming over the next two decades is quite similar across the SSPs due to competing effects of warming (methane, ozone) and cooling (aerosols) SLCFs. For the scenarios with the most stringent climate and air pollution mitigations (SSP1-1.9 and SSP1-2.6), the likely near-term warming from the SLCFs is predominantly due to sulphate aerosol reduction, but this effect levels off after 2040. In the absence of climate change policies and with weak air pollution control (SSP3-7.0), the likely near-term warming due to changes in SLCFs is predominantly due to increases in methane, ozone and HFCs, with smaller contributions from changes in aerosols. SSP5-8.5has the highest SLCF-induced warming rates due to warming from methane and ozone increases and reduced aerosols due to stronger air pollution control compared to the SSP3-7.0scenario. {6.7.2}
At the end of the century, the large diversity of GSAT response to SLCF changes among the scenarios robustly covers the possible futures, as the scenarios are internally consistent and span a range from very high to very low emissions. In the scenarios without climate change mitigation (SSP3-7.0 and SSP5-8.5) the likely range of the estimated warming due to SLCFs in 2100 relative to 2019 is 0.4°C–0.9°C {6.7.3, 6.7.4}. InSSP3-7.0there is a near-linear warming due to SLCFs of 0.08°C per decade, while for SSP5-8.5there is a more rapid warming in the first half of the century. For the scenarios considering the most stringent climate and air pollution mitigations (SSP1-1.9 and SSP1-2.6), the reduced warming from reductions in methane, ozone and HFCs partly balances the warming from reduced aerosols, and the overall SLCF effect is a likely increase in GSAT of 0.0°C–0.3°C in 2100, relative to 2019. The SSP2-4.5scenario (with moderate climate change and air pollution mitigations) results in a likely warming of 0.2°C–0.5°C in 2100 due to SLCFs, with the largest warming from reductions in aerosols. {6.7.3}
Potential Effects of SLCF Mitigation
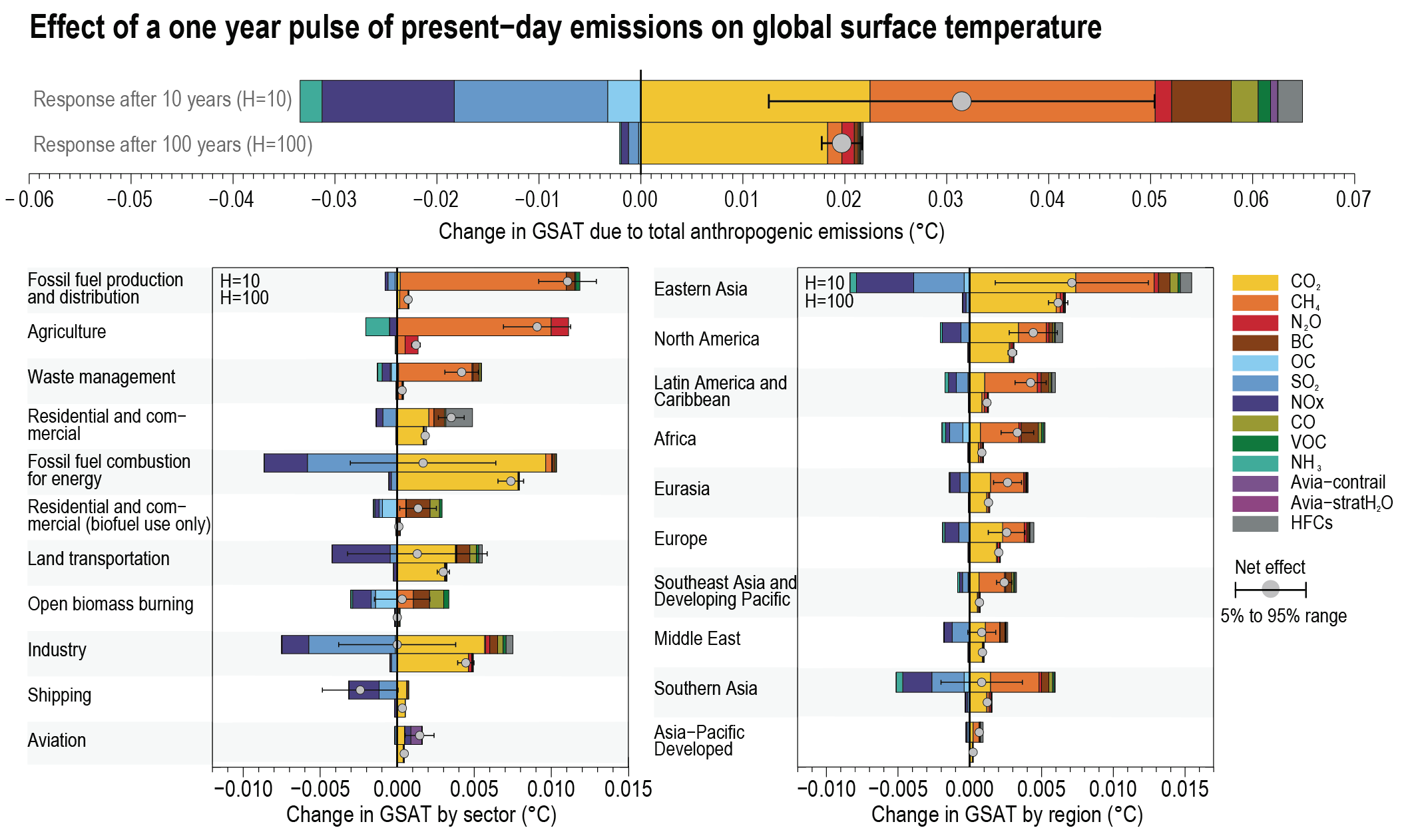
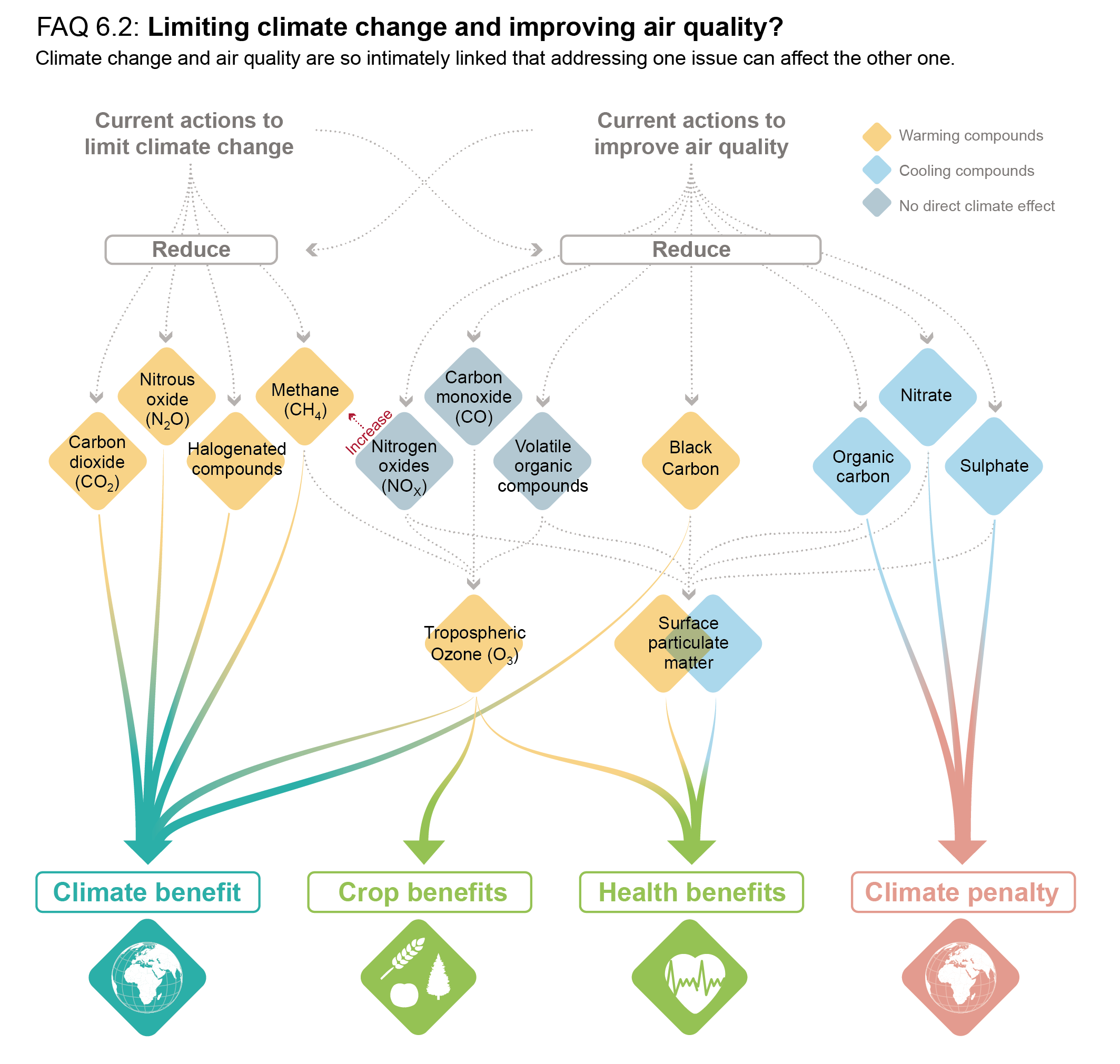
Over time scales of 10 to 20 years, the global temperature response to a year’s worth of current emissions of SLCFs is at least as large as that due to a year’s worth of CO2 emissions (high confidence). Sectors producing the largest SLCF-induced warming are those dominated by methane emissions: fossil fuel production and distribution, agriculture and waste management (high confidence). On these time scales, SLCFs with cooling effects can significantly mask the CO2 warming in the case of fossil fuel combustion for energy and land transportation, or completely offset the CO2 warming and lead to an overall net cooling in the case of industry and maritime shipping (prior to the implementation of the revised fuel-sulphur limit policy for shipping in 2020) (medium confidence). Ten years after a one-year pulse of present-day aviation emissions, SLCFs induce strong but short-lived warming contributions to the GSAT response (medium confidence), while CO2 both gives a warming effect in the near term and dominates the long-term warming impact (high-confidence). {6.6.1, 6.6.2}
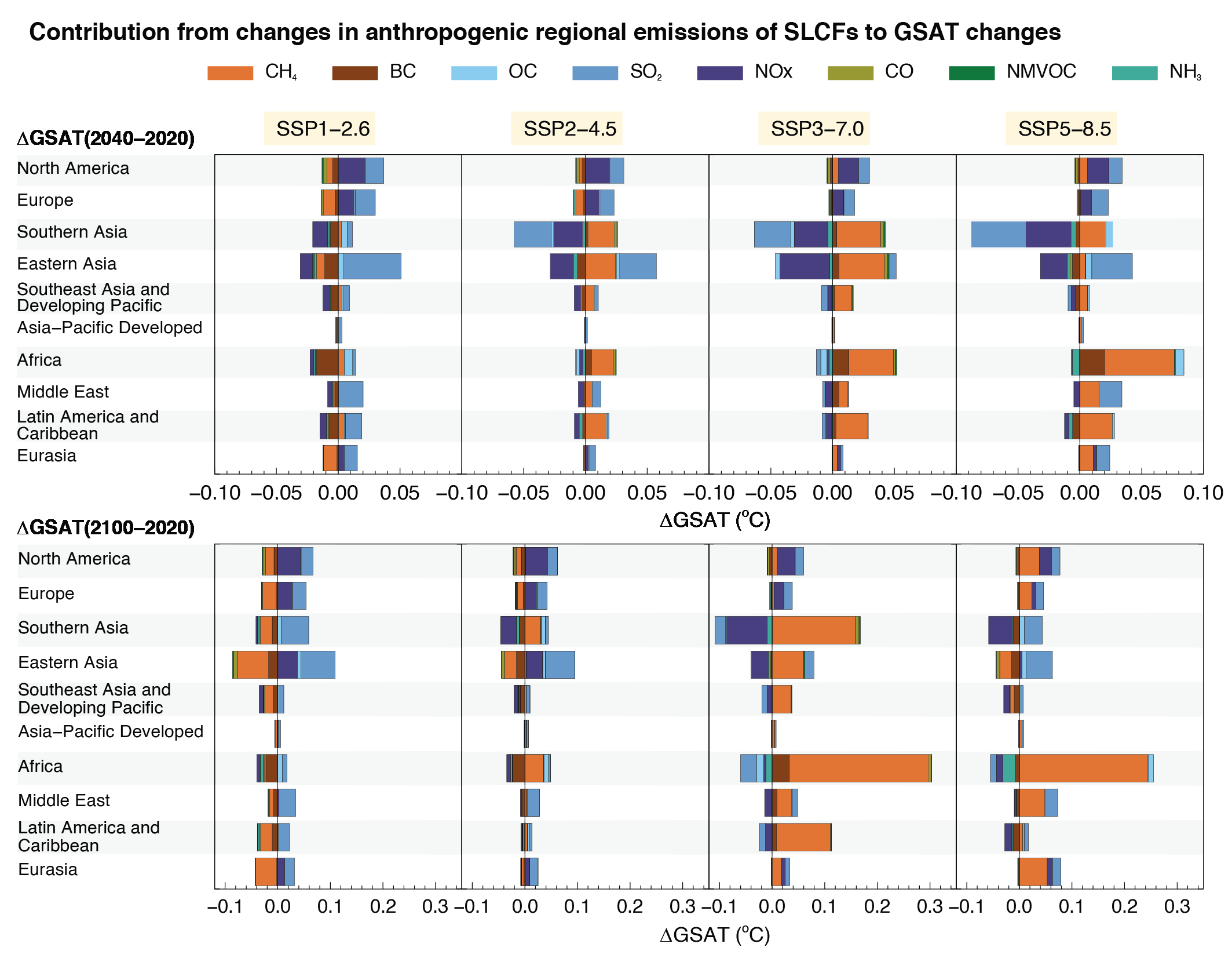
The effects of SLCFs decay rapidly over the first few decades after pulse emission. Consequently, on time scales longer than about 30 years, the net long-term global temperature effects of sectors and regions are dominated by CO2 (high confidence). The global mean temperature response following a climate change mitigation measure that affects emissions of both short- and long-lived climate forcers depends on their atmospheric decay times, how fast and for how long the emissions are reduced, and the inertia in the climate system. For SLCFs including methane, the rate of emissions drives the long-term global temperature effect, as opposed to CO2 for which the long-term global temperature effect is controlled by the cumulative emissions. About 30 years or more after a one-year emission pulse occurs, the sectors contributing the most to global warming are industry, fossil fuel combustion for energy and land transportation, essentially through CO2 (high confidence). Current emissions of SLCFs, CO2 and N2O from Eastern Asia and North America are the largest regional contributors to additional net future warming on both short (medium confidence) and long time scales (high confidence). {6.6.1, 6.6.2}
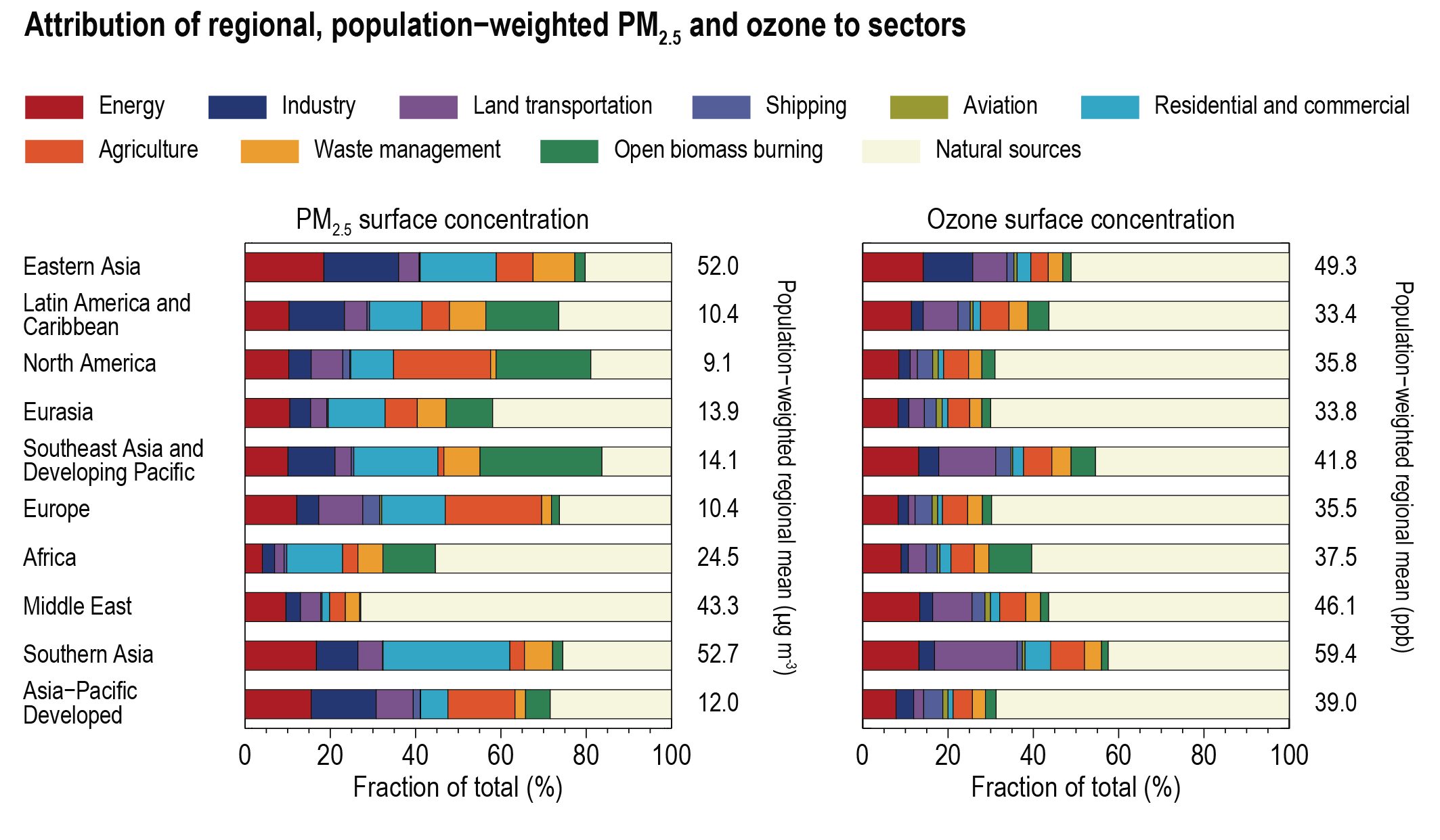
At present, emissions from the residential and commercial sectors (fossil and biofuel use for cooking and heating) and the energy sector (fossil fuel production, distribution and combustion) contribute the most to the world population’s exposure to anthropogenic fine PM (high confidence), whereas emissions from the energy and land transportation sectors contribute the most to ozone exposure (medium tohigh confidence). The contribution of different sectors to PM varies across regions, with the residential sector being the most important in Southern Asia and Africa, agricultural emissions dominating in Europe and North America, and industry and energy production dominating in Central and Eastern Asia, Latin America and the Middle East. Energy and industry are important PM2.5 contributors in most regions, except Africa (high confidence). Sector contributions to surface ozone concentrations are similar for all regions. {6.6.2}
Assuming implementation and efficient enforcement of both the Kigali Amendment to the Montreal Protocol on Ozone Depleting Substances and current national plans to limit emissions (as in SSP1-2.6), the effects of HFCs on GSAT, relative to 2019, would remain below +0.02°C from 2050 onwards versus about +0.04°C to +0.08°C in 2050 and +0.1°C to +0.3°C in 2100 considering only national HFC regulations decided prior to the Kigali Amendment (as inSSP5-8.5) (medium confidence). Further improvements in the efficiency of refrigeration and air-conditioning equipment during the transition to low-global-warming-potential refrigerants would bring additional greenhouse gas reductions (medium confidence) resulting in benefits for climate change mitigation and to a lesser extent for air quality due to reduced air pollutant emissions from power plants. {6.6.3, 6.7.3}
Future changes in SLCFs are expected to cause additional warming. This warming is stable after 2040 in scenarios leading to lower global air pollution as long as methane emissions are also mitigated, but the overall warming induced by SLCF changes is higher in scenarios in which air quality continues to deteriorate (induced by growing fossil fuel use and limited air pollution control) (high confidence). If a strong air pollution control resulting in reductions in anthropogenic aerosols and non-methane ozone precursors was considered in SSP3-7.0, it would lead to a likely additional near-term global warming of 0.08 [0.00 to 0.10] °C in 2040. An additional concomitant methane mitigation (consistent with SSP1’s stringent climate change mitigation policy implemented in the SSP3 world) would not only alleviate this warming but would turn this into a cooling of 0.07°C with a likely range of [–0.02 to +0.14]°C (compared with SSP3-7.0 in 2040) . Across the SSPs, the collective reduction of methane, ozone precursors and HFCs can make a difference of 0.2°C with a very likely range of [0.1 to 0.4] °C in 2040 and 0.8°C with a very likely range of [0.5 to 1.3] °C at the end of the 21st century (comparing SSP3-7.0 and SSP1-1.9), which is substantial in the context of the Paris Agreement. Sustained methane mitigation, wherever it occurs, stands out as an option that combines near- and long-term gains on surface temperature (high confidence) and leads to air-quality benefits by reducing surface ozone levels globally (high confidence). {6.6.3, 6.7.3, 4.4.4}
Rapid decarbonization strategies lead to air-quality improvements but are not sufficient to achieve, in the near term, air-quality guidelines set for fine PM by the World Health Organization (WHO), especially in parts of Asia and in some other highly polluted regions (high confidence). Additional methane and BC mitigation would contribute to offsetting the additional warming associated with SO2 reductions that would accompany decarbonization (high confidence). Strong air pollution control as well as strong climate change mitigation, implemented separately, lead to large reductions in exposure to air pollution by the end of the century (high confidence). Implementation of air pollution controls, relying on the deployment of existing technologies, leads more rapidly to air quality benefits than climate change mitigation (high confidence), which requires systemic changes. However, in both cases, significant parts of the population are projected to remain exposed to air pollution exceeding the WHO guidelines (high confidence). Additional policies envisaged to attain Sustainable Development Goals (SDGs; e.g., access to clean energy, waste management) bring complementary SLCF reduction. Only strategies integrating climate, air quality, and development goals are found to effectively achieve multiple benefits. {6.6.3, 6.7.3, Box 6.2}
Implications of COVID-19 Restrictions for Emissions, Air Quality and Climate
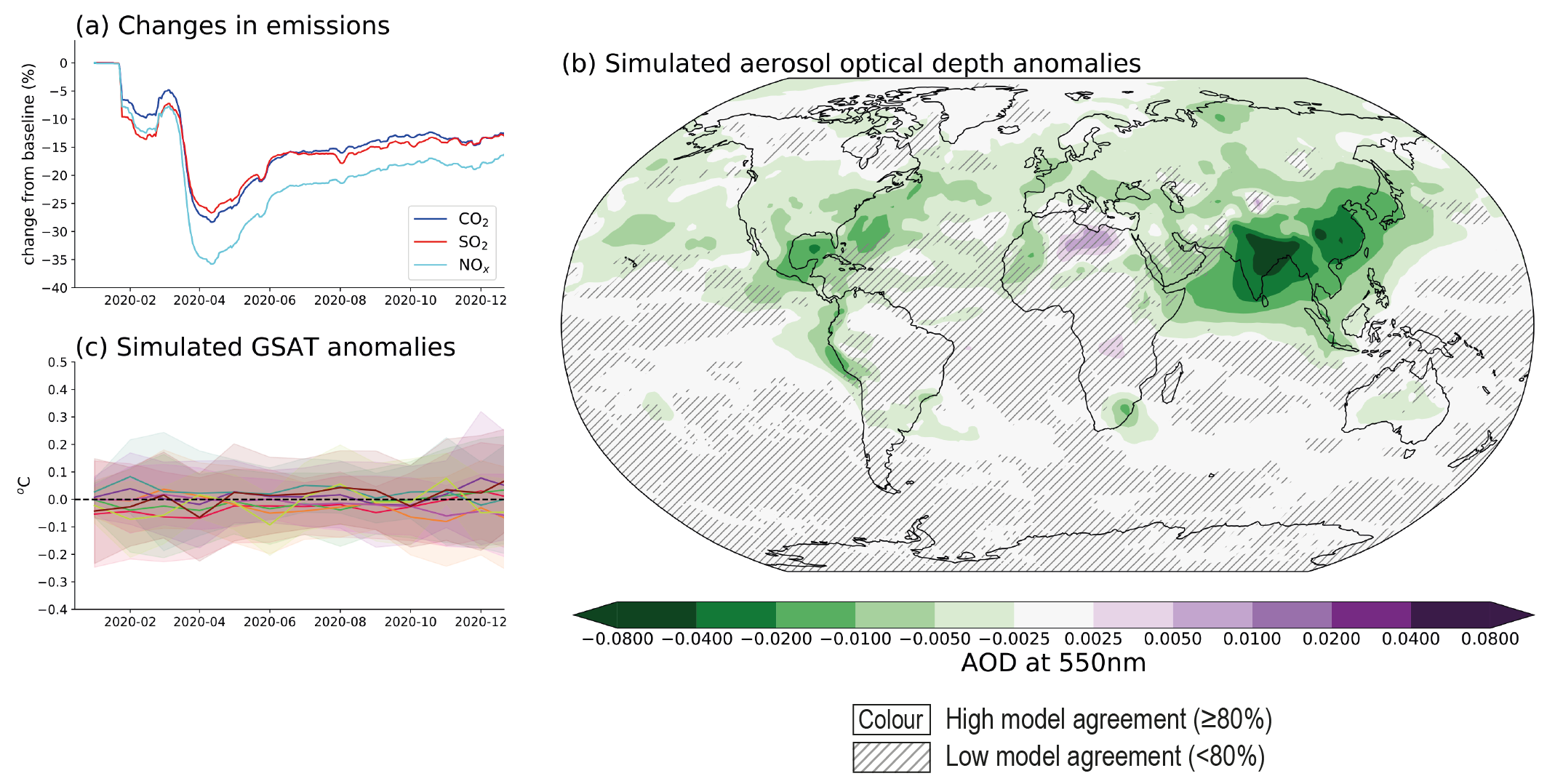
Emissions reductions associated with COVID-19 containment led to a discernible temporary improvement of air quality in most regions, but changes to global and regional climate are undetectable above internal variability. Global anthropogenic NOx emissions decreased by a maximum of about 35% in April 2020 (medium confidence). There ishigh confidence that, with the exception of surface ozone, these emissions reductions have contributed to improved air quality in most regions of the world. Global fossil CO2 emissions decreased by 7% (with a range of 5.8–13.0%) in 2020 relative to 2019, largely due to reduced emissions from the transportation sector (medium confidence). Overall, the net ERF, relative to ongoing trends, from COVID-19 restrictions was likely small and positive for 2020 (less than 0.2 W m–2), thus temporarily adding to the total anthropogenic climate influence, with positive forcing from aerosol changes dominating over negative forcings from CO2, NOx and contrail cirrus changes. Consistent with this small net radiative forcing, and against a large component of internal variability, Earth system model simulations show no detectable effect on global or regional surface temperature or precipitation (high confidence). {Cross-Chapter Box 6.1}
6.1 Introduction
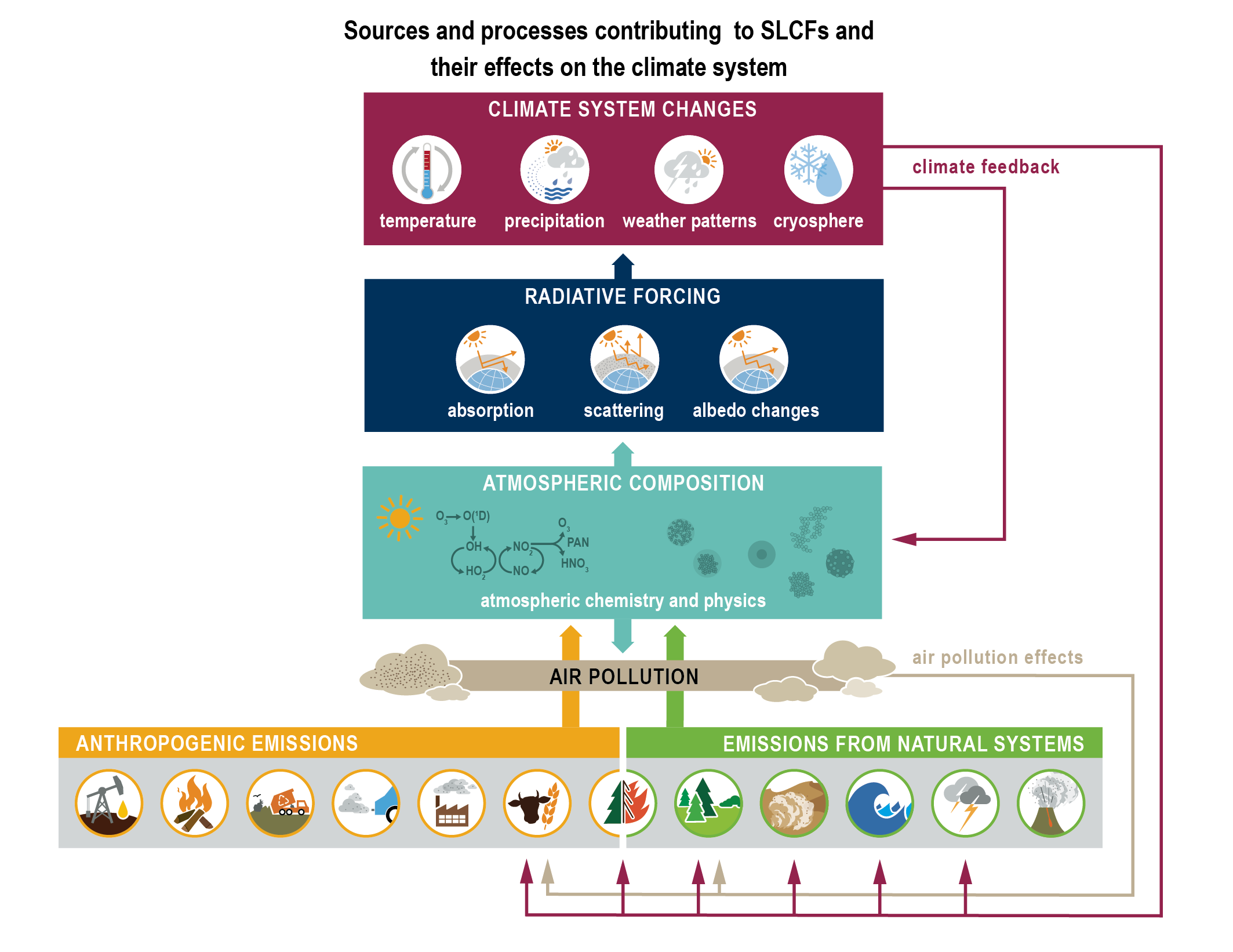
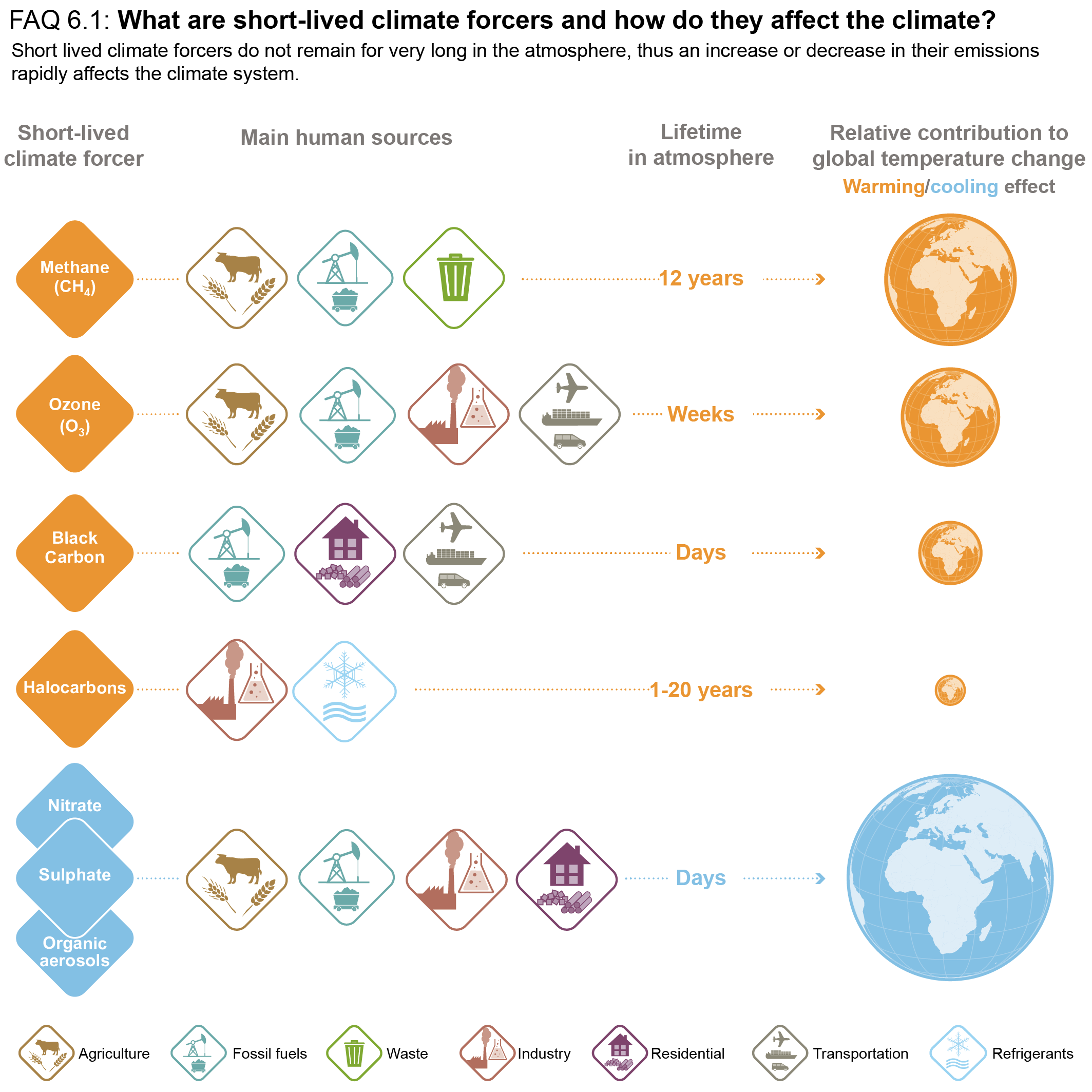
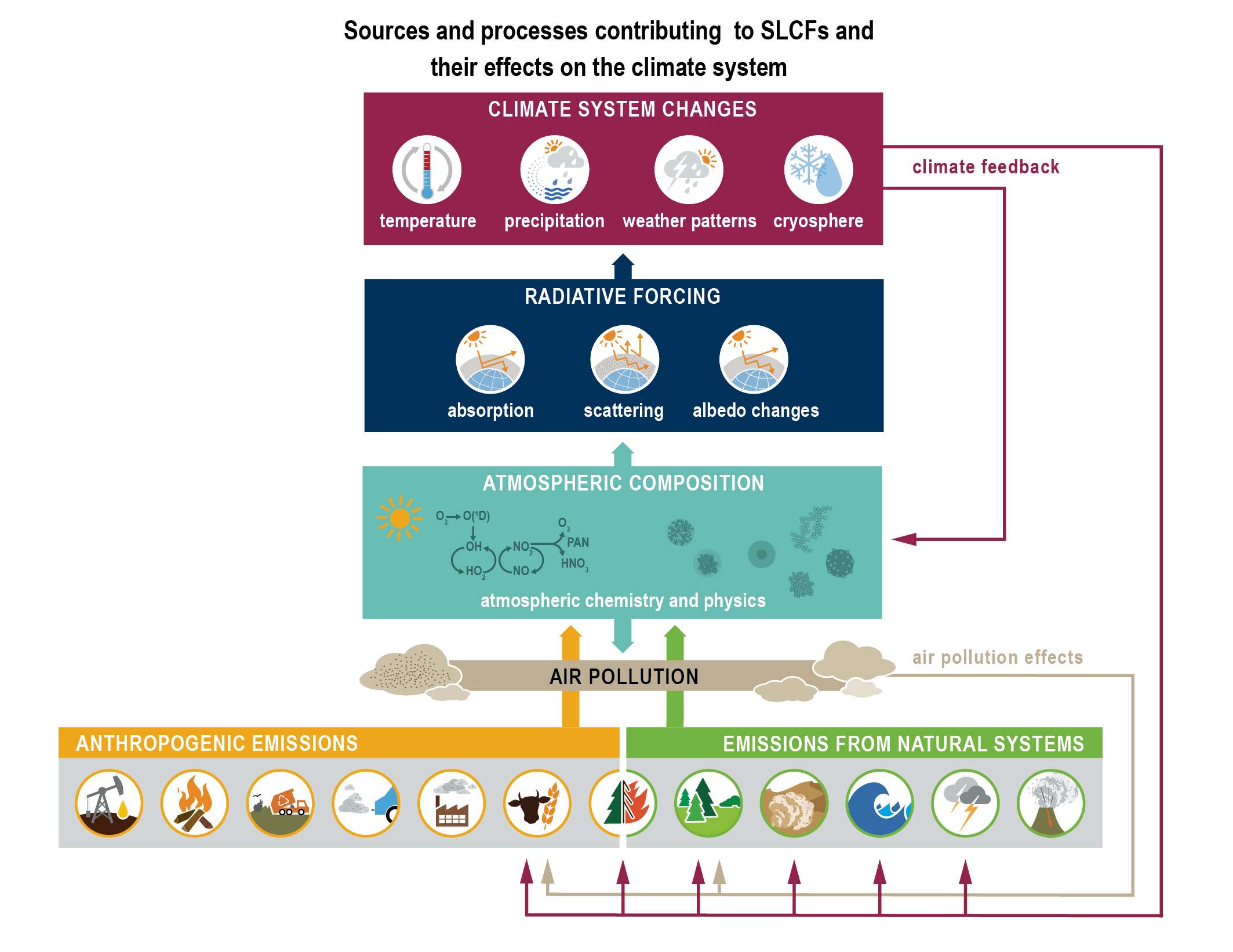
Short-lived climate forcers (SLCFs) are a set of chemically and physically reactive compounds with atmospheric lifetimes typically shorter than two decades but differing in terms of physiochemical properties and environmental effects. SLCFs can be classified as direct or indirect, with direct SLCFs exerting climate effects through their radiative forcing and indirect SLCFs being precursors of direct climate forcers. Direct SLCFs include methane (CH4), ozone (O3), short-lived halogenated compounds, such as hydrofluorocarbons (HFCs), hydrochlorofluorocarbons (HCFCs), and aerosols. Indirect SLCFs include nitrogen oxides (NOx), carbon monoxide (CO), non-methane volatile organic compounds (NMVOCs), sulphur dioxide (SO2), and ammonia (NH3). Aerosols consist of sulphate (SO24–), nitrate (NO–3), ammonium (NH+4), carbonaceous aerosols (e.g., black carbon (BC), organic aerosols (OA)), mineral dust, and sea spray (see Table 6.1) and can be present as internal or external mixtures and at sizes from nano-meters to tens of micro-meters. SLCFs can be emitted directly from natural systems and anthropogenic sources (primary) or can be formed by reactions in the atmosphere (secondary; Figure 6.1).
6.1.1 Importance of SLCFs for Climate and Air Quality
The atmospheric lifetime determines the spatial and temporal variability, with most SLCFs showing high variability, except methane and many HCFCs and HFCs that are also well-mixed (as a consequence methane is discussed together with other well-mixed greenhouse gases (GHGs) in Chapters 2, 5, and 7). In contrast to well-mixed GHGs, such as CO2, methane and some HFCs, the radiative forcing effects of most SLCFs are largest at regional scales and climate effects predominantly occur in the first two decades after their emissions or formation. However, changes in their emissions can also induce long-term climate effects, for instance by altering some biogeochemical cycles. Therefore, the temporal evolution of radiative effects of SLCFs follows that of emissions, that is, when SLCF emissions decline to zero their atmospheric abundance and radiative effects decline towards zero. The total influence of individual SLCF emissions on radiative forcing and climate includes their effects on the abundances of other forcers through chemistry (chemical adjustments).
SLCFs can affect climate by interacting with radiation or by perturbing other components of the climate system (e.g., the cryosphere and carbon cycle through deposition, or the water cycle through modifications of cloud properties via cloud condensation nuclei or ice nuclei). SLCFs can have either net warming or net cooling effects on climate. In addition to altering the Earth’s radiative balance, many SLCFs are also air pollutants with adverse effects on human health and ecosystems. SLCFs are of interest for climate policies (e.g., methane, HFCs), and are regulated as air pollutants (e.g., aerosols, ozone) or because of their deleterious influence on stratospheric ozone (e.g., HCFCs). The list of SLCFs assessed in this chapter and their effects are provided in Table 6.1.
Compounds | Source Type | Lifetime | Direct | Indirect | Climate Forcing | Other Effects on Climatea | WHO AQ Guidelinesb |
CH4 | Primary | ~9 years ~12 years (perturbation time) | CH4 | O3, H2O, CO2 | + | Noc | |
O3 | Secondary | Hours to weeks | O3 | CH4, secondary organic and sulphate aerosols | + | Ecosystems | 100 μg m–3 8-hour mean |
NOx(= NO + NO2) | Primary | Hours to days | O3, nitrate aerosols, CH4 | +/– | Ecosystems | 40 μg m–3 annual mean200 μg m–3 1-hour mean | |
CO | Primary + Secondary | 1 to 4 months | O3, CH4 | + | No | ||
NMVOCs** | Primary + Secondary | Hours to months | O3, CH4, organic aerosols | +/– | No | ||
SO2 | Primary | Days (trop.) to weeks (strat.) | Sulphate and nitrate aerosols, O3 | – | Ecosystems | 20 μg m–3 24-hour mean500 μg m–3 10-minute mean | |
NH3 | Primary | Hours | Ammonium Sulphate, Ammonium Nitrate | – | Ecosystems | No | |
HCFCs | Primary | Months to years | HCFCs | O3 | +/– | Noc | |
HFCs | Primary | Days to years | HFCs | + | Noc | ||
Halons and Methylbromide | Primary | Years | Halons and Methylbromide | Stratospheric O3 | +/– | Noc | |
Very Short-lived Halogenated Species (VSLSs) | Primary | Less than 6 months | O3 | – | Noc | ||
Sulphate aerosols | Secondary | Minutes to weeks | Sulphate | – | Clouds Ecosystems | as part of PMd | |
Nitrate aerosols | Secondary | Minutes to weeks | Nitrate | – | Clouds Ecosystems | as part of PMd | |
Carbonaceous Aerosols | Primary + Secondary | Minutes to Weeks | BC, OA | +/– | Cryo, Clouds Ecosystems | as part of PMd | |
Sea spray | Primary | Day to week | Sea spray | – | Clouds Ecosystems | as part of PMd | |
Mineral dust | Primary | Minutes to Weeks | Mineral dust | +/– | Cryo Cloud Ecosystems | as part of PMd |
* for lifetimes reported in this table, it is assumed that the compounds are uniformly mixed throughout the troposphere, however, this assumption is unlikely for compounds with lifetimes <1 year and, therefore, the reported values should be viewed as approximations (Prather et al., 2001).
** Some NMVOCs are biogenic volatile organic compounds (BVOCs).
aClouds: effect on clouds through aerosol–cloud interactions, Ecosystems: effect on ecosystems through changes in radiation and deposition, Cryo: effect on planetary albedo through deposition on snow and ice;bKrzyzanowski and Cohen (2008);cregulated through Kyoto/Montreal protocols;d for Particulate Matter with diameter <2.5 µm (PM2.5): 10 µg m–3annual mean or 25 µg m–324-hour mean (99th percentile) and for Particulate Matter with diameter <10 µm (PM10): 20 µg m–3annual mean or 50 µg m–324-hour mean (99th percentile).
As depicted in Figure 6.1, emissions of SLCFs are governed by anthropogenic activities and sources from natural systems (see Section 6.2 for details). Atmospheric chemistry in this context is both a source and a sink of SLCFs. For instance, ozone and secondary aerosols are exclusively formed through atmospheric mechanisms (Sections 6.3.2 and 6.3.5 respectively). The hydroxyl (OH) radical, the most important oxidizing agent in the troposphere, acts as a sink for SLCFs by reacting with them and thereby influencing their lifetime (Section 6.3.6). Through SLCF radiative forcing and feedbacks (Section 6.4), key climate parameters, such as temperature, hydrological cycle and weather patterns are perturbed. Climate change also influences air quality (Section 6.5). As depicted in Figure 6.1, SLCFs affect both climate and air quality, hence SLCF mitigation has linkages to both issues (Section 6.6). Socio-economic narratives including air-quality policies determine future projections of SLCFs in the five core Shared Socio-economic Pathways (SSPs): SSP1-1.9, SSP1-2.6, SSP2-4.5, SSP3-7.0, and SSP5-8.5 (described in Chapter 1), and in addition, a subset of SSP3 scenarios make it possible to isolate the effect of various SLCF mitigation trajectories on climate and air quality (Section 6.7).
6.1.2 Treatment of SLCFs in Previous Assessments
Although ozone, aerosols and their precursors have been considered in previous IPCC assessment reports, AR5 considered SLCFs as a specific category of climate-relevant compounds but referred to them as near-term climate forcers (NTCFs; Myhre et al. , 2013). In AR5, the linkages between air quality and climate change were also considered in a more detailed and quantitative way than in previous reports (Kirtman et al., 2013; Myhre et al., 2013).
The AR5 WGI assessed radiative forcings for short-lived gases, aerosols, aerosol precursors and aerosol–cloud interactions as well as the evolution of confidence levels in the forcing mechanisms from SAR to AR5. Whereas the forcing mechanisms for ozone and aerosol–radiation interactions were estimated to be characterized with high confidence, the ones induced by aerosols through other processes remained of very low to low confidence. The AR5 also reported that forcing agents such as aerosols and ozone are highly heterogeneous spatially and temporally, and these patterns affect global and regional temperature responses as well as other aspects of climate response such as the hydrologic cycle (Myhre et al. , 2013).
The AR5 WGI also evaluated the air quality-climate interaction through the projected trends of surface ozone and PM2.5. Kirtman et al. (2013) concluded with high confidence that the response of air quality to climate-driven changes is more uncertain than the response to emissions-driven changes, and also that locally higher surface temperatures in polluted regions will trigger regional feedbacks in chemistry and local emissions that will increase peak levels of ozone and PM2.5 (medium confidence).
In the IPCC Special Report on Global Warming of 1.5°C (SR1.5; Allen et al., 2018a), Rogelj et al. (2018a) state that the evolution of methane and SO2 emissions strongly influences the chances of limiting warming to 1.5°C, and that, considering mitigation scenarios to limit warming to 1.5°C or 2°C, a weakening of aerosol cooling would add to future warming in the near term, but can be tempered by reductions in methane emissions (high confidence). In addition, as some SLCFs are co-emitted alongside CO2, especially in the energy and transport sectors, low CO2 scenarios, relying on decline of fossil fuel use, can result in strong abatement of some cooling and warming SLCFs (Rogelj et al., 2018a). On the other hand, specific reductions of the warming SLCFs (methane and BC) would, in the short term, contribute significantly to the efforts of limiting warming to 1.5°C. Reductions of BC and methane would have substantial co-benefits, improving air quality and therefore limiting effects on human health and agricultural yields. This would, in turn, enhance the institutional and socio-cultural feasibility of such actions in line with the United Nations’ Sustainable Development Goals (SDGs; Coninck et al., 2018).
Following SR1.5, the IPCC Special Report on Climate Change and Land (SRCCL; IPCC, 2019a) took into consideration the emissions on land of three major SLCFs: mineral dust, carbonaceous aerosols (BC and OA) and biogenic volatile compounds (BVOCs) (Jia et al., 2019). The SRCCL concluded that: (i) there is no agreement about the direction of future changes in mineral dust emissions; (ii) fossil fuel and biomass burning, and secondary organic aerosols (SOA) from natural BVOC emissions are the main global sources of carbonaceous aerosols whose emissions are expected to increase in the near future due to possible increases in open biomass burning and increase in SOA from oxidation of BVOCs (medium confidence); and (iii) BVOCs are emitted in large amounts by forests and they are rapidly oxidized in the atmosphere to form less volatile compounds that can condense and form SOA, and in a warming planet, BVOC emissions are expected to increase but magnitude is unknown and will depend on future land-use change, in addition to climate (limited evidence, medium agreement).
Finally, the IPCC Special Report on the Ocean and Cryosphere in a Changing Climate (SROCC; IPCC, 2019b) discussed the effects of BC deposition on snow and glaciers, concluding that there is high confidence that darkening of snow through the deposition of BC and other light-absorbing particles enhances snowmelt in the Arctic (Meredith et al., 2019), but that there is limited evidence and low agreement that long-term changes in glacier mass of high mountain areas are linked to light-absorbing particles (Hock et al., 2019).
6.1.3 Chapter Roadmap
Figure 6.2 presents the Chapter 6 roadmap.
Specific aspects of SLCFs can also be found in other chapters of this report: the evolution of ozone, HFCs and aerosols, as well as the long-term evolution of methane, dust and volcanic aerosols are discussed in Chapter 2; near-term climate projections and SLCFs are discussed in Chapter 4; the global budget of methane is addressed in Chapter 5; aerosol–cloud and aerosol–precipitation interactions are treated in Chapters 7 and 8, respectively; the global radiative forcing of SLCFs is assessed in Chapter 7; some aspects of downscaling methodology in climate modelling concerning SLCFs are discussed in Chapter 10. The WGII report assesses how climate change affects air pollution and its impacts on human health and the WGIII report assesses the role of SLCFs in abatement strategies and their cost-effectiveness, the implications of mitigation efforts on air pollution as well as the articulation between air pollution policies and GHG mitigation.
This chapter discusses air quality from a global point of view with a focus on surface ozone and particulate matter concentrations. Local and indoor air pollution, as well as the effect of air pollution on health, are beyond the scope of this chapter. This Assessment is mainly based on results and studies relying on global models or observation datasets operated through global networks or from satellites. Global chemistry-climate modelsenable the quantification of changes in background concentrations, such as changes in surface ozone due to large-scale changes in climate or methane, by considering comprehensively the physiochemical processes (Box 6.1). In addition, climate effects are often non-linear responses to concentrations which already respond non-linearly to emissions, with per-mass unit effects often larger in pristine than in polluted regions, justifying the relevance of global models. However, specific aspects of urban air quality cannot be captured by global models and require high-resolution models that reproduce the temporal and spatial variability of emissions and abundances necessary to precisely account for the non-linearity of the chemistry and the sensitivity of local air pollution to its drivers. Consequently, the sectoral analysis in Section 6.6 and the mitigation effects in Section 6.7 cannot be directly applied for local air-quality planning.
Due to their short lifetimes, SLCF trends and effects are strongly related to the localization and evolution of the emissions sources. To better link the drivers of emissions evolution and SLCFs, Chapter 6 makes use of regions defined by the WGIII in most of the analysis. An exception is made for the effect of SLCFs on the climate, for which analysis relies on WGI Atlas regions.
6.2 Global and Regional Temporal Evolution of SLCF Emissions
SLCF emissions originate from a variety of sources driven by anthropogenic activities and natural processes. The natural sources include vegetation, soil, fire, lightning, volcanoes and oceans. Changes in SLCF emissions from natural systems occur either due to human activities, such as land-use change, or due to global changes. Their sensitivity to climate change thus induces climate feedbacks (see Section 6.4.5 for a quantification of these feedbacks). This section reviews the current understanding of historical emissions for anthropogenic, natural, and open biomass burning sources. A detailed discussion of methane sources, sinks, trends are provided in Chapter 5, Section 5.2.2.
6.2.1 Anthropogenic Sources
Estimates of global anthropogenic (human-caused) SLCF emissions and their historical evolution that were used in AR5 (CMIP5; Lamarque et al., 2010) have been revised for use in CMIP6 (Hoesly et al., 2018). The update considered new data and assessment of the impact of environmental policies, primarily regarding air pollution control (R. Wang et al. , 2014; S.X. Wang et al. , 2014; Montzka et al. , 2015; Crippa et al. , 2016; Turnock et al. , 2016; Klimont et al. , 2017a; Zanatta et al. , 2017; Prinn et al. , 2018). Additionally, Hoesly et al. (2018) have extended estimates of anthropogenic emissions back to 1750 and developed an updated and new set of spatial proxies allowing for more differentiated (source sector-wise) gridding of emissions (Feng et al., 2020). The CMIP6 emissions inventory has been developed with the Community Emissions Data System (CEDS) that improves upon existing inventories with a more consistent and reproducible methodology, similar to approaches used in, for example, the EDGAR database (Crippa et al., 2016) and the GAINS model (Amann et al., 2011; Klimont et al., 2017a; Höglund-Isaksson et al., 2020) where emissions of all compounds are consistently estimated using the same emissions drivers and propagating individual components (activity data and emissions factors) separately to capture fuel and technology trends affecting emissions trajectories over time. This contrasts with the approach used to establish historical emissions for CMIP5 where different datasets available at the time were combined. The CMIP6 exercise is based on the first release of the CEDS emissions dataset (version 2017-05-18, sometimes referred to hereafter as CMIP6 emissions) whose main features regarding SLCFs are described hereafter. The CEDS has been and will be regularly updated and extended; the recent update of the CEDS (Hoesly et al., 2019) and consequences for this Assessment is discussed when necessary. Some details on how SLCF emissions have been represented in scenarios used by IPCC assessments can be found in Chapter 1 (Section 1.6.1 and Cross-Chapter Box 1.4 and in Section 6.7.1.1).
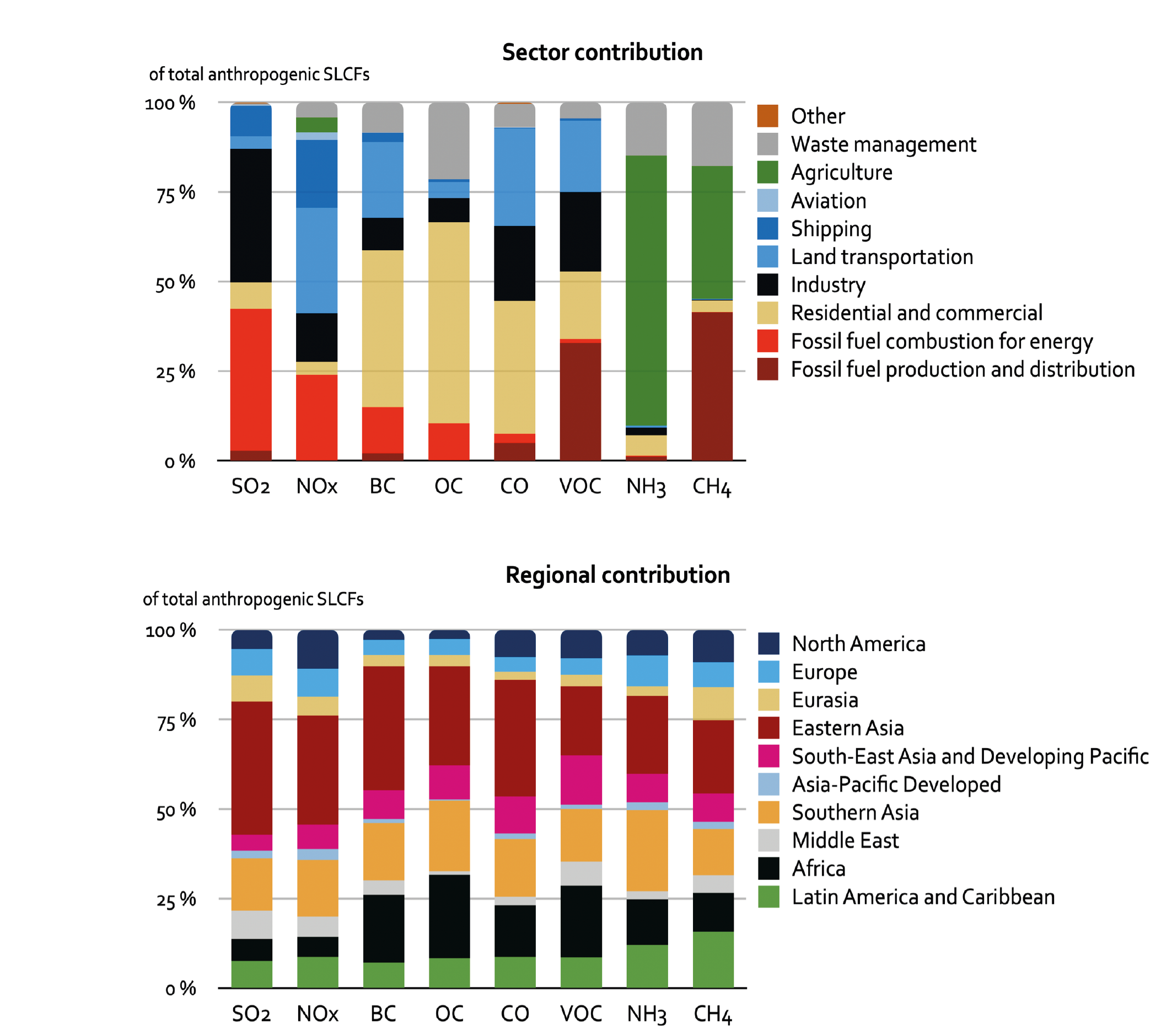
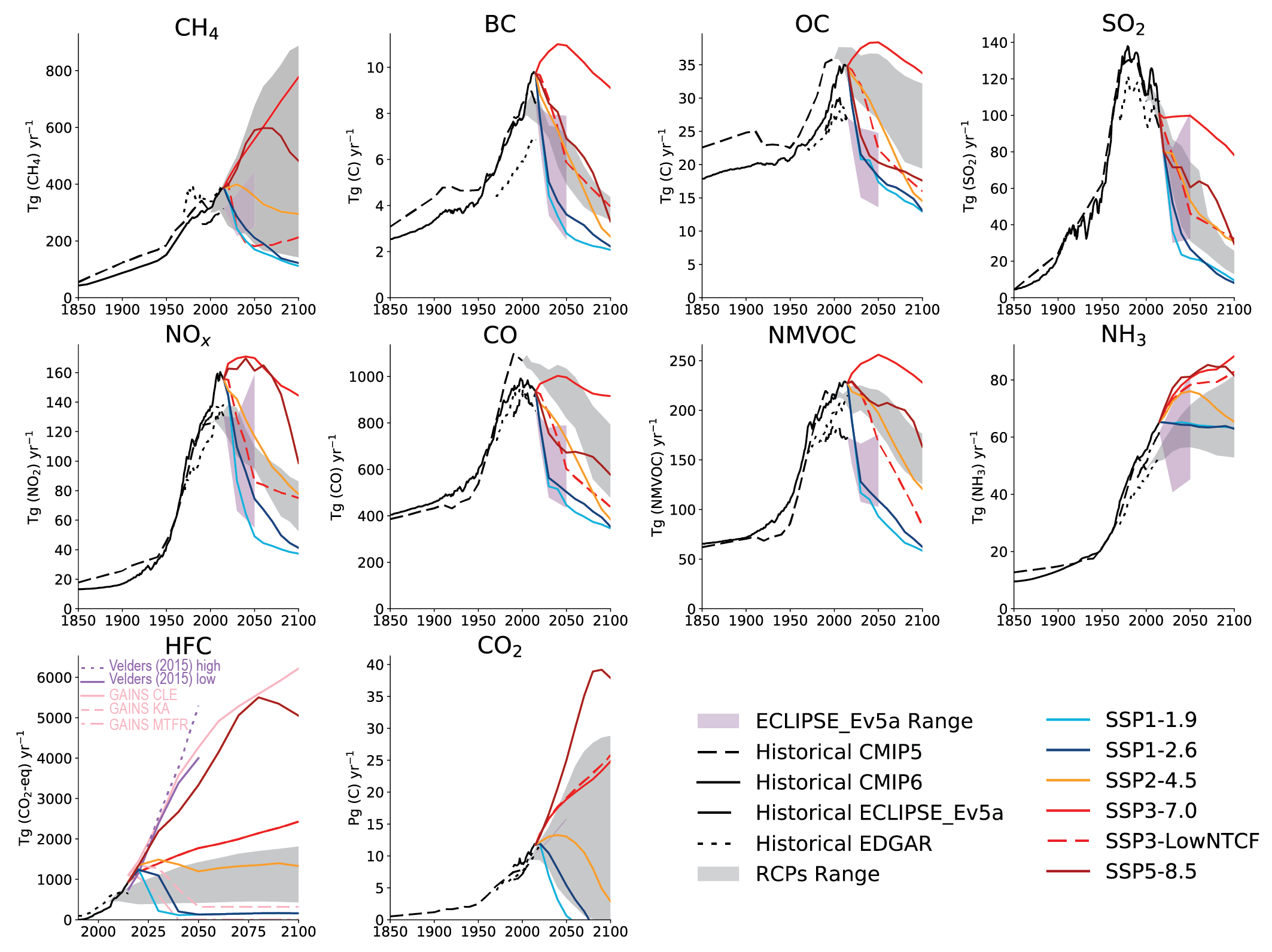
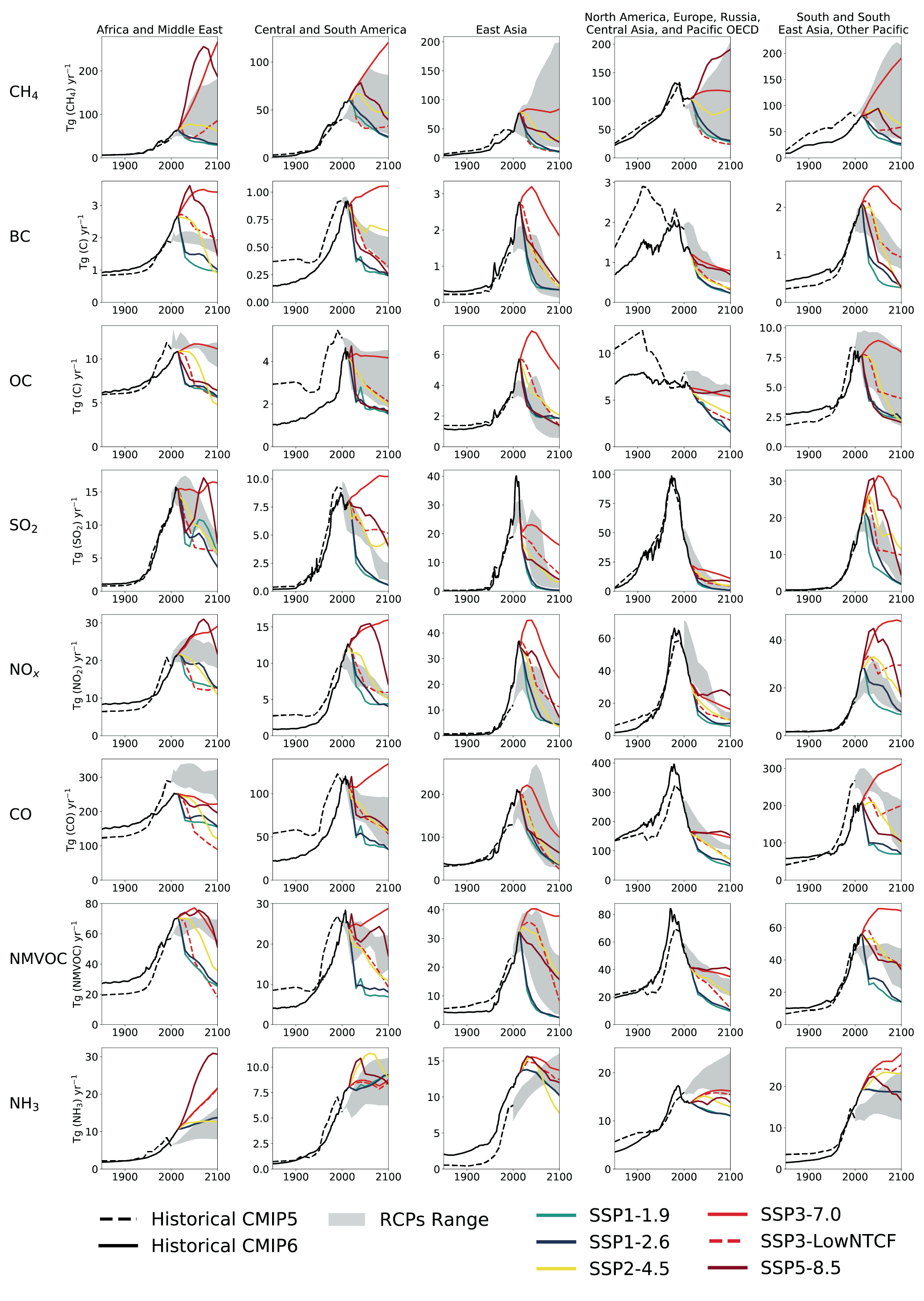
For most of the SLCF species, the global and regional anthropogenic emissions trends developed for CMIP6 for the period 1850–2000 are not substantially different from those used in CMIP5 (Figures 6.18 and 6.19) despite the different method used to derive them. Hoesly et al. (2018, CEDS) developed independent time series capturing trends in fuel use, technology and level of control, whereas CMIP5 combined different emissions datasets. However, for the period after 1990, the CMIP6 dataset shows for all species, except for SO2, CO, and (since 2011) for NOx, a different trend than CMIP5 (i.e., continued strong growth of emissions driven primarily by developments in Asia (Figure 6.19)). The unprecedented growth of emissions from Eastern and Southern Asia since 2000 changed the global landscape of emissions, making Asia the dominant SLCF source region (Figures 6.3 and 6.19). The Representative Concentration Pathways (RCP) scenarios used in AR5 started from the year 2000 (van Vuuren et al., 2011) and did not capture the SLCF emissions which actually occurred until 2015. The CEDS inventory (Hoesly et al., 2018) includes improved representation of these trends and the estimate for 2014. These findings have been largely supported by several independent emissions inventory studies and remote-sensing data analysis. However, for the last decade the decline of Asian emissions of SO2 and NOx appears underestimated while growth of BC and OC emissions in Asia and Africa seems overestimated in CMIP6, compared to most recent regional evaluations (Klimont et al., 2017a; Zheng et al., 2018b; Elguindi et al., 2020; Kanaya et al., 2020; McDuffie et al., 2020), which are largely considered in the updated release of the CEDS (Hoesly et al., 2019). Consequently, global CMIP6 anthropogenic emissions for 2014 are likely overestimated by about 10% for SO2 and NOx and by about 15% for BC and OC.
For SO2, independent emissions inventories and observational evidence show that on a global scale strong growth of Asian emissions has been countered by reduction in North America and Europe (Reis et al. , 2012; Amann et al. , 2013; Crippa et al. , 2016; Aas et al. , 2019). However, Chinese emissions declined by nearly 70% between about 2006 and 2017(high confidence) (Silver et al. , 2018; Zheng et al. , 2018b; Mortier et al. , 2020; Tong et al. , 2020). The estimated reduction in China contrasts with continuing strong growth of SO2 emissions in Southern Asia (Figure 6.19). In 2014, over 80% of anthropogenic SO2 emissions originated from power plants and industry, with Asian sources contributing more than 50% of the total (Figure 6.3).
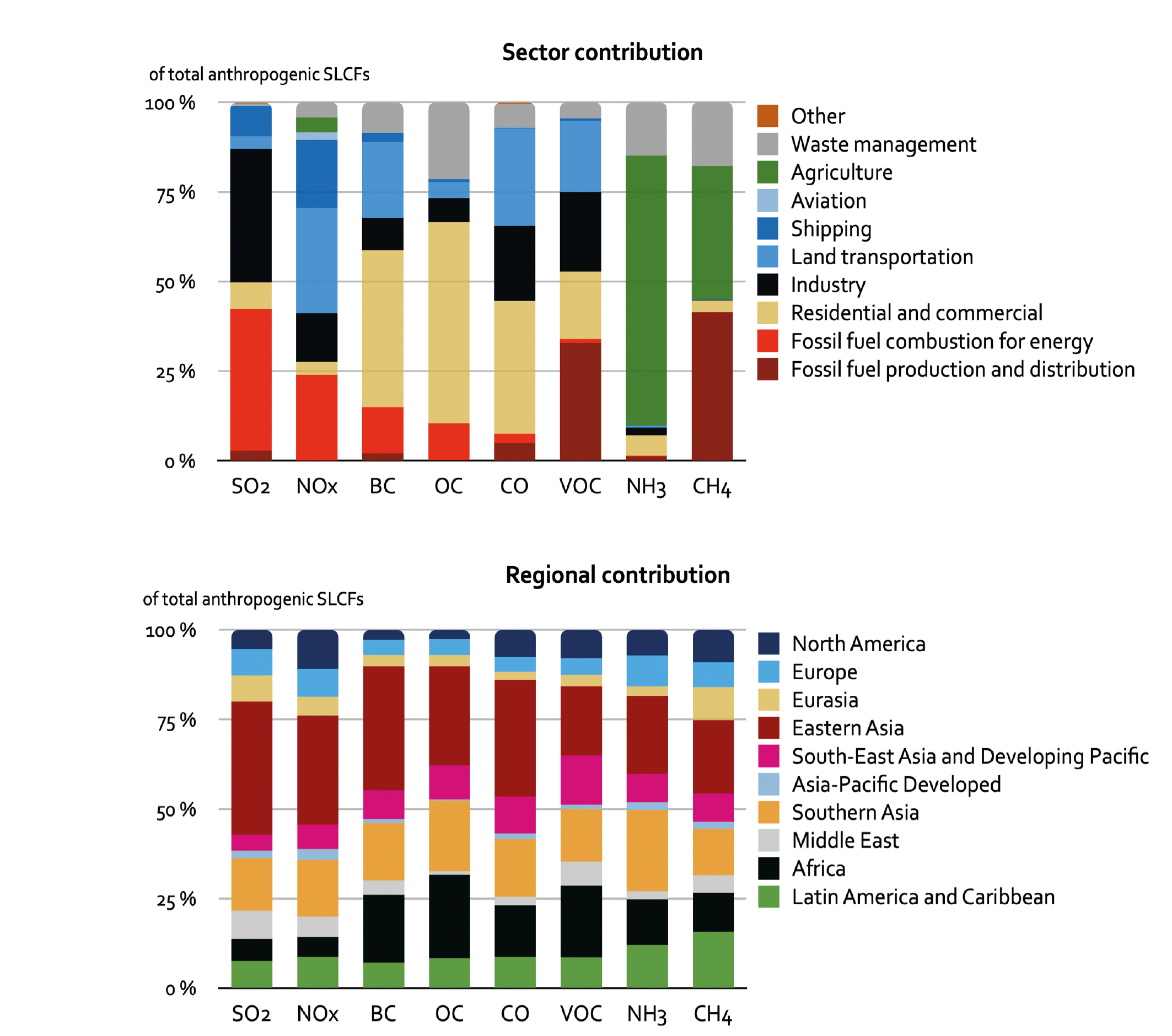
Global emissions of NOx have been growing in spite of the successful reduction of emissions in North America, Europe, Japan and Korea (Crippa et al., 2016; Turnock et al., 2016; Miyazaki et al., 2017; Jiang et al., 2018), partly driven by continuous efforts to strengthen the emissions standards for road vehicles in most countries (Figures 6.18 and 6.19). In many regions, an increase in vehicle fleet as well as non-compliance with emissions standards (Anenberg et al. , 2017, 2019; Jonson et al. , 2017; Jiang et al. , 2018), growing aviation (Grewe et al., 2019; Lee et al., 2021) and demand for energy, and consequently a large number of new fossil fuel power plants, have more than compensated for these reductions. Since about 2011, global NOx emissions appear to have stabilized or slightly declined (medium confidence) but the global rate of decline has been underestimated in the CEDS, as recent data suggest that emissions reductions in China were larger than included in the CEDS (Figure 6.19 and Hoesly et al., 2018). Recent bottom-up emissions estimates (Zheng et al., 2018b) largely confirm what has been shown in satellite data (F. Liu et al., 2016; Miyazaki et al., 2017; Silver et al., 2018): a strong decline of NO2 column over eastern China (high confidence) (Section 6.3.3.1). At a global level, the estimated CEDS CO emissions trends are comparable to NOx, which has been confirmed by several inverse modelling studies (Section 6.3.3.2). The transport sector (including international shipping and aviation) was the largest anthropogenic source of NOx (about 50% of the total) and also contributed over 25% of CO emissions in 2014; Asia represented 50% and North America and Europe about 20% of global total NOx and CO emissions (Figure 6.3).
Oil production-distribution and transport sectors have dominated anthropogenic NMVOC emissions for most of the 20th century (Hoesly et al., 2018) and still represent a large share (Figure 6.3). Efforts to control transport emissions (i.e., increasing stringency of vehicle emissions limits) were largely offset by the fast growth of emissions from chemical industries and solvent use, as well as from fossil fuel production and distribution, resulting in continued growth of global anthropogenic NMVOC emissions since 1900 (high confidence) (Figure 6.18). Since AR5, there is high confidence that motor vehicle NMVOC emissions have sharply declined in North America and Europe in the last decades (Rossabi and Helmig, 2018), for example, by about an order of magnitude in major US cities since 1990 (Bishop and Haugen, 2018; McDonald et al., 2018). Increasing (since 2008) oil- and gas-extraction activities in North America lead to a strong growth of NMVOC emissions (high confidence) as shown by analysis of ethane column data (Franco et al., 2016), but absolute emission amounts remain uncertain (Pétron et al., 2014; Tzompa-Sosa et al., 2019). In Eastern Asia, there is medium confidence in a decreasing trend of motor vehicle emissions, suggested by ambient measurements in Beijing since 2002 (Wang et al., 2015) and by bottom-up estimates (Zheng et al., 2018b), and a decrease in residential heating emissions due to declining coal and biofuel use since 2005 (Zheng et al., 2018b; M Li et al., 2019). However, total anthropogenic NMVOC emissions have increased steadily in China since the mid-20th century, largely due to the growing importance of the solvent-use and industrial sectors (medium evidence, high agreement) (Sun et al. , 2018; Zheng et al. , 2018b; M. Li et al. , 2019). Resulting changes in the NMVOC speciated emissions might be underestimated in the current regional and global inventories. For example, in the USA, a recent study suggested an emergent shift in urban NMVOC sources from transportation to chemical products (i.e., household chemicals, personal care products, solvents, etc.), which is not in accordance with emissions inventories currently used (McDonald et al., 2018). In many European regions and cities, wood burning has been increasingly used for residential heating, partly for economic reasons and because it is considered CO2-neutral (Athanasopoulou et al., 2017); in situ measurements in several cities, including Paris, suggest that wood burning explains up to half of the NMVOC emissions during winter (Kaltsonoudis et al., 2016; Languille et al., 2020). Due to the vast heterogeneity of sources and components of NMVOCs, uncertainty in regional emissions and trends is higher than for most other components.
Emissions of carbonaceous aerosols (BC, OC) have been steadily increasing and their emissions have almost doubled since 1950 (medium confidence) (Hoesly et al., 2018). Before 1950, North America and Europe contributed about half of the global total but successful introduction of diesel particulate filters on road vehicles (Fiebig et al., 2014; Robinson et al., 2015; Klimont et al., 2017a) and declining reliance on solid fuels for heating brought in large reductions (high confidence) (Figure 6.19). Currently, global carbonaceous aerosol emissions originate primarily from Asia and Africa (Bond et al., 2013; Hoesly et al., 2018; Elguindi et al., 2020; McDuffie et al., 2020), representing about 80% of the global total (high confidence) (Figure 6.3). Consideration, in CMIP6, of emissions from kerosene lamps and gas flaring, revised estimates for open burning of waste, regional coal consumption, and new estimates for Russia (Stohl et al., 2013; Huang et al., 2015; Huang and Fu, 2016; Kholod et al., 2016; Conrad and Johnson, 2017; Evans et al., 2017; Klimont et al., 2017a) resulted in over 15% higher global emissions of OC and BC than in the CMIP5 estimates for the first decade of the 21st century (Figure 6.18). However, the continued increase of BC emissions over Eastern Asia after 2005, estimated in CMIP6 (Figure 6.19), has been questioned recently as a steady decline of BC concentrations was measured in the air masses flowing out from the east coast of China (Kanaya et al., 2020), which has been also estimated in recent regional bottom-up and top-down inventories (Zheng et al. , 2018a; Elguindi et al. , 2020; McDuffie et al. , 2020). Since AR5, confidence in emissions estimates and trends in North America and Europe has increased, but high uncertainties remain for Asia and Africa, despite their major contribution to global emissions. The size distribution of emitted species, of importance for climate and health impacts, remains uncertain and the CEDS inventory does not provide such information. Overall, a factor two uncertainty in global estimates of BC and OC emissions remains, with post-2005 emissions overestimated in Asia (high confidence) and Africa (medium confidence).
Bottom-up global emissions estimates of methane (Lamarque et al., 2010; Hoesly et al., 2018; Janssens-Maenhout et al., 2019; Höglund-Isaksson et al., 2020) for the last two decades are higher than top-down assessments (e.g., Saunois et al., 2016, 2020) but trends from the two methods are similar and indicate continued growth (high confidence). Larger discrepancies exist at the sectoral and regional levels, notably for coal mining (Peng et al., 2016; Miller et al., 2019) and the oil and gas sector due to the growth of unconventional production and higher loss estimates Section 5.2.2; Franco et al. , 2016; Alvarez et al. , 2018; Dalsøren et al. , 2018).
Agricultural production (livestock and mineral nitrogen fertilizer application) is the primary source of ammonia in the atmosphere with more than half of present-day emissions originating in Asia (Hoesly et al. , 2018; Figure 6.3, EC-JRC/PBL, 2020; Vira et al. , 2020). NH3 emissions are estimated to have grown strongly since 1850, especially since 1950, driven by continuously increasing livestock production, widespread application of mineral nitrogen fertilizers, and lack of action to control ammonia (high confidence) (Erisman et al. , 2008; Riddick et al. , 2016; Hoesly et al. , 2018; Fowler et al. , 2020). The trends estimated in CMIP5 and CMIP6 are similar, while in absolute terms, CMIP6 has somewhat higher emissions as it includes emissions from wastewater and human waste that were largely missing in CMIP5 (Hoesly et al., 2018). CMIP6 has improved spatial and temporal distribution of emissions (Lamarque et al., 2013a) relying on the EDGAR v4.3 database and Paulot et al. (2014), but important uncertainties remain for regionally specific temporal patterns (Riddick et al. , 2016; Liu et al. , 2019; Feng et al. , 2020; Vira et al. , 2020). The continuing increase in global NH3 emissions is driven primarily by growing livestock and crop production in Asia while emissions in the USA and Europe remain about constant or have slightly declined in the last decade (Hoesly et al., 2018). Recent satellite and ground observations support trends estimated in CMIP6 dataset (Section 6.3.3.4).
To summarize, there are significant differences in spatial and temporal patterns of SLCF emissions across global regions (Figure 6.18). Until the 1950s, the majority of SLCF emissions were associated with fossil fuel use (SO2, NOx, NMVOCs, CO) and about half of BC and OC originated from North America and Europe (Lamarque et al., 2010; Hoesly et al., 2018). Since the 1990s a large redistribution of emissions was associated with strong economic growth in Asia and declining emissions in North America and Europe due to air-quality legislation and the declining capacity of energy-intensive industry; currently more than 50% of anthropogenic emissions of each SLCF species (including methane and NH3) originates from Asia (Figure 6.3; Amann et al. , 2013; Bond et al. , 2013; Fiore et al. , 2015; Crippa et al. , 2016, 2018; Klimont et al. , 2017a; Hoesly et al. , 2018). The dominance of Asia for SLCF emissions is corroborated by growing remote-sensing capacity that has been providing an independent evaluation of estimated pollution trends in the last decade (Duncan et al. , 2013; Lamsal et al. , 2015; Luo et al. , 2015; Fioletov et al. , 2016; Geddes et al. , 2016; Irie et al. , 2016; Krotkov et al. , 2016; Wen et al. , 2018).
Since AR5, the quality and completeness of activity and emission-factor data and applied methodology, including spatial allocation together with independent satellite-derived observations, have improved, raising confidence in methods used to derive emissions. There is high confidence in the sign of global trends of SLCF emissions until the year 2000. However, only medium confidence for the rate of change in the two last decades, owing primarily to uncertainties in the actual application of reduction technologies in fast-growing economies of Asia. At a regional level, bottom-up derived SLCF emissions trends, and magnitudes in regions with strong economic growth and changing air-quality regulation, are highly uncertain but can be better constrained with top-down methods (Section 6.3). For most SLCF species, there is high confidence in trends and magnitudes for affluent countries from the Organisation for Economic Co- operation and Development (OECD) regions where accurate and detailed information about drivers of emissions exists; medium confidence is assessed for regional emissions of NH3, methane and NMVOC.
6.2.2 Emissions by Natural Systems
This section assesses our current understanding of SLCF emissions by natural systems. Many naturally occurring emission processes in the Earth system have been perturbed by the growing influence of human activities either directly (e.g., deforestation, agriculture) or via human-induced atmospheric CO2 increase and climate change, and therefore cannot be considered as purely natural emissions. The temporal evolution and spatial distribution of natural SLCF emissions are highly variable and their estimates rely on models with rather uncertain parametrizations for production mechanisms. For some SLCFs, the natural processes by which emissions occur are also not well understood. In the following sections, we assess the level of confidence in present-day SLCF emissions by natural systems, in their perturbation since the pre-industrial period and their sensitivity to future changes. When available, the assessment also includes estimates from the CMIP6 model ensemble. Note that volcanic SO2 emissions are discussed in Section 2.2.2 and natural sources of methane and N2O are assessed in Sections 5.2.2 and 5.2.3.
6.2.2.1 Lightning NOx
Lightning contributes approximately 10% of the total NOx emissions (Murray, 2016). Since lightning NOx (LNOx) is predominantly released in the upper troposphere, it has a disproportionately large impact on ozone and OH, and on the lifetime of methanecompared with surface NOx emissions. Whereas the global spatial and temporal distribution of lightning flashes can be characterized thanks to satellite-borne and ground sensors (Virts et al., 2013; Cecil et al., 2014), constraining the amount of NOx produced per flash (Miyazaki et al. , 2014; Medici et al. , 2017; Nault et al. , 2017; Marais et al. , 2018; D.J. Allen et al. , 2019; Bucsela et al. , 2019) and its vertical allocation (Koshak et al., 2014; Medici et al., 2017) has been more elusive.
Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP) models in CMIP5 used a range of LNOx4 between 1.2–9.7 TgN y−1 (Lamarque et al., 2013b). In CMIP6, the corresponding LNOx range is between 3.2–7.6 TgN y−1 (Griffiths et al., 2021). All CMIP6 models (as well as most models included in CMIP5, Young et al., 2013) apply a parametrization that relates cloud-top height to lightning intensity (Price and Rind, 1992), projecting an increase in LNOx in a warmer world in the range of 0.27–0.61 TgN yr−1°C–1 (Thornhill et al., 2021a). However, models using parametrizations based on convection (Grewe et al., 2001), updraft mass flux (Allen and Pickering, 2002) or ice flux (Finney et al., 2016a) show either much less sensitivity or a negative response (Finney et al., 2016b, 2018; Clark et al., 2017).
In summary, the total present-day global lightning NOx emissions are still estimated to be within a factor of two. There is high confidence that LNOx are perturbed by climate change; however, there is low confidence in the sign of the change due to fundamental uncertainties in parametrizations.
6.2.2.2 NOx emissions from Soils
Soil NOx (SNOx) emissions occur in connection with complex biogenic/microbial nitrification and denitrification processes (Ciais et al., 2013), which in turn are sensitive – in a non-linear manner – to temperature, precipitation, soil moisture, carbon and nutrient content, and the biome itself (e.g., Hudman et al., 2012). Global SNOx estimates, based on observationally constrained chemistry-transport model and vegetation model studies, show a broad range between 4.7–16.8 TgN yr–1 (Young et al., 2018). This estimate is generally larger than the current source strength used in CMIP6 simulations, which is prescribed using an early empirical estimate, typically scaled to about 5 TgN yr–1 (Yienger and Levy, 1995).
By the end of the 21st century, the overall nitrogen fixation in non-agricultural ecosystems could be 40% larger than in 2000, due to increased enzyme activity with growing temperatures, but the emission rates of NO (and N2O) could be dominated by changes in precipitation patterns and evapotranspiration fluxes (Fowler et al., 2015). Current Earth system models (ESMs) incorporate biophysical and biogeochemical processes only to a limited extent (Jia et al., 2019), precluding adequate climate sensitivity studies for SNOx. Hence, while the current strength source of soil NOx has been better constrained over the last decade, adequate representations of SNOx and how it escapes from the canopy, which could provide quantitative estimates of climate-driven changes in SNOx, are still missing in ESMs.
6.2.2.3 Vegetation Emissions of Organic Compounds
A wide range of BVOCs are emitted from vegetation with the dominant compounds being isoprene and monoterpenes but also including sesquiterpenes, alkenes, alcohols, aldehydes and ketones. The photooxidation of BVOC emissions plays a fundamental role in atmospheric composition by controlling the regional and global budgets of ozone and organic aerosols, and impacting the lifetime of methane and other reactive components (Arneth et al., 2010b; Heald and Spracklen, 2015). Substantial uncertainty exists across different modelling frameworks for estimates of global total BVOC emissions and individual compound emissions (Messina et al., 2016). Global isoprene emissions estimates differ by a factor of two from 300–600 TgC yr–1 and global monoterpene emissions estimates by a factor of five from 30–150 TgC yr–1 (Messina et al., 2016). A main driver of the uncertainty ranges is the choice of basal emissions rates assigned to different plant functional types in the model; however, the smaller uncertainty range for isoprene than for monoterpenes is not fully understood (Arneth et al., 2008). The evaluation of global BVOC emissions is challenging because of poor measurement data coverage in many regions and the lack of year-round measurements (Unger et al., 2013). Several observational approaches have been developed in the past few years to improve understanding of BVOC emissions, including indirect methods such as the measurement of the OH loss rate in forested environments (Yang et al., 2016) and application of the variability in satellite formaldehyde concentrations (Palmer et al., 2006; Barkley et al., 2013; Stavrakou et al., 2014). Direct space-borne isoprene retrievals using infrared radiance (IR) measurements have very recently become available (Fu et al., 2019; Wells et al., 2020). Collectively these approaches have identified weaknesses in the ability of the parametrizations in global models to reproduce BVOC emissions hotspots (Wells et al., 2020). However, none of the current observational approaches have yet been able to reduce the uncertainty ranges in global emissions estimates.
At the plant level, BVOC emissions rates and composition depend strongly on plant species with plants tending to emit either isoprene or monoterpenes but not both. Photosynthetic activity is a main driver of isoprene and monoterpene production. Therefore, radiation and temperature, along with leaf-water status, phenological state and atmospheric CO2 mixing ratio, affect emissions directly (on the leaf scale) and indirectly (via plant productivity; Guenther et al. , 2012; Loreto et al. , 2014; Niinemets et al. , 2014). CO2 directly influences the isoprene-synthesis process, with inhibition under increasing atmospheric CO2 (Rosenstiel et al., 2003; Possell et al., 2005; Wilkinson et al., 2009). Direct CO2 inhibition has been observed for some monoterpene compounds (Loreto et al., 2001; Llorens et al., 2009). Severe/long-term water stress may reduce emissions whilst mild/short-term water stress may temporarily amplify or maintain BVOC emissions to protect plants against ongoing stress (Peñuelas and Staudt, 2010; Potosnak et al., 2014; Genard-Zielinski et al., 2018). Furthermore, observations in the Amazon indicate that the chemical composition of monoterpene emissions could also change under elevated temperature conditions (Jardine et al., 2016). In addition, all these processes are investigated over short time scales but the long-term response of BVOC emissions depends on how the vegetation itself responds to the altered climate state (including temperature and water stress).
Global BVOC emissions are highly sensitive to environmental changes including changes in climate, atmospheric CO2, and vegetation composition and cover changes in natural and managed lands. Recent global modelling studies agree that global isoprene emissions have declined since the pre-industrial period, driven predominantly by anthropogenic land-use and land-cover change (LULCC) with results converging on a 10–25% loss of isoprene emissions between 1850 and the present day (Lathière et al., 2010; Unger, 2013, 2014; Acosta Navarro et al., 2014; Heald and Geddes, 2016; Hantson et al., 2017; Hollaway et al., 2017; Scott et al., 2017). The historical evolution of monoterpene and sesquiterpene emissions is less well studied and there is no robust consensus on even the sign of the change (Acosta Navarro et al., 2014; Hantson et al., 2017). Future global isoprene and monoterpene emissions depend strongly on the climate and land-use scenarios considered (Hantson et al., 2017; Szogs et al., 2017). BVOC emissions will be sensitive to future land-based climate change mitigation strategies including afforestation and bioenergy, with impacts of bioenergy depending on the choice of crops (Szogs et al., 2017).
Most CMIP6 models use overly simplistic parametrizations and project an increase in global BVOC emissions in response to warming temperatures (Turnock et al., 2020). This good agreement actually reflects the lack of diversity in BVOC-emissions parametrizations in global models that do not fully account for the complex processes influencing emissions that are discussed above.
Overall, we assess that historical global isoprene emissions declined between the pre-industrial period and the present day by 10–25% (low confidence) but historical changes in global monoterpenes and sesquiterpenes are too uncertain to provide an assessment. Future changes in BVOCs depend strongly on the evolution of climate and land use and are strongly sensitive to land-based climate change mitigation strategies. However, the net response of BVOC emissions is uncertain due to the complexity of processes that are hard to constrain observationally and are considered with various degrees of details in models.
6.2.2.4 Land Emissions of Dust Particles
The emission of dust particles into the atmosphere results from a natural process, namely saltation bombardment of the soil by large wind-blown particles, such as sand grains, and from disintegration of saltating particle clusters (Kok et al., 2012). The occurrence and intensity of dust emissions are controlled by soil properties, vegetation and near-surface wind, making dust emissions sensitive to climate change and LULCC (Jia et al., 2019). In addition, dust can be directly emitted through human activities, such as agriculture, off-road vehicles, building construction and mining, and indirectly emitted through hydrological changes due to human actions such as water diversion for irrigation (e.g., Ginoux et al., 2012). Estimates of the anthropogenic fraction of global dust vary from less than 10% to over 60% suggesting that the human contribution to the global dust budget is quite uncertain (Ginoux et al., 2012; Stanelle et al., 2014; Xi and Sokolik, 2016). Reconstruction of global dust (deposition) from paleo records indicate factor of two to four changes between the different climate regimes in the glacial and interglacial periods (Section 2.2.6).
An extremely limited number of studies have explored the evolution of global dust sources since pre-industrial times (Mahowald et al., 2010; Stanelle et al., 2014). A modelling study estimated a 25% increase in global dust emissions between the late 19th century and the present, due to agricultural land expansion and climate change (Stanelle et al., 2014). CMIP5 models were unable to capture the observed variability of annual and longer time scales in North African dust emissions (Evan et al., 2014), however, more recent ESMs with process-based dust emissions schemes that account for changes in vegetation and climate in a more consistent manner, better match the observations (Kok et al., 2014; Evans et al., 2016). Feedbacks between the global dust cycle and the climate system (Section 6.4.5) could account for a substantial fraction of the total aerosol feedbacks in the climate system with an order of magnitude enhancement on a regional scale (Kok et al., 2018). In summary, there is high confidence that atmospheric dust source and loading are sensitive to changes in climate and land use, however, there is low confidence in quantitative estimates of dust emission response to climate change.
6.2.2.5 Oceanic Emissions of Marine Aerosols and Precursors
Oceans are a significant source of marine aerosols that influence climate directly by scattering and absorbing solar radiation or indirectly through the formation of cloud condensation nuclei (CCN) and ice nucleating particles (INPs). Marine aerosols consist of primary sea-spray particles and secondary aerosols produced by the oxidation of emitted precursors, such as dimethylsulphide (DMS) and numerous other BVOCs. Sea-spray particles, composed of sea salt and primary organic aerosols (POA), are produced by wind-induced wave breaking as well as the direct mechanical disruption of waves. The understanding of sea-spray emissions has increased substantially over the last five years, however, the knowledge of formation pathways and factors influencing their emissions continue to have large uncertainties (Forestieri et al., 2018; Saliba et al., 2019). The emission rate of sea-spray particles is predominantly controlled by wind speed. Since AR5, the influence of other factors, including sea surface temperature, wave history and salinity is increasingly evident (Callaghan et al. , 2014; Grythe et al. , 2014; Ovadnevaite et al. , 2014; Salter et al. , 2014; Barthel et al. , 2019). Marine POA, often the dominant submicron component of sea spray, are emitted as a result of oceanic biological activity, however the biological processes by which these particles are produced remain poorly characterized contributing to large uncertainties in global marine POA emissions estimates (Tsigaridis et al. , 2014; Cravigan et al. , 2020; Hodzic et al. , 2020) . Furthermore, the particle size and chemical composition of sea-spray particles, and how these evolve in response to changing climate factors and dynamic oceanic biology, continue to have large uncertainties.
DMS, the largest natural source of sulphur in the atmosphere, is produced by marine phytoplankton and is transferred from ocean water to the atmosphere due to wind-induced mixing of surface water. DMS oxidizes to produce sulphate aerosols and contributes to the formation of CCN. Since AR5, the range in global DMS flux estimates reduced from 10–40 TgS yr–1 to 9–34 TgS yr–1 with a very likely range of 18–24 TgS yr–1based on sea-surface measurements and satellite observations (Lana et al., 2011). DMS production, and consequently emissions, have been shown to respond to multiple stressors, including climate warming, eutrophication, and ocean acidification. However, large uncertainties in process-based understanding of the mechanisms controlling DMS emissions, from physiological to ecological, limit our knowledge of past variations and our capacity to project future changes.
Overall, there is low confidence in the magnitude and changes in marine aerosol emissions in response to shifts in climate and marine ecosystem processes.
6.2.2.6 Open Biomass Burning Emissions
Emissions from open biomass burning (including forest, grassland, peat fires and agricultural waste burning) represent about 30%, 10%, 15% and 40% of present-day global emissions of CO, NOx, BC and OC, respectively (van Marle et al., 2017; Hoesly et al., 2018). Wildfires also play an important role in several atmospheric chemistry–climate feedback mechanisms (Bowman et al., 2009; Fiore et al., 2012) and fire events occurring near populated areas induce severe air pollution episodes (Marlier et al., 2020; Rooney et al., 2020; Yu et al., 2020).
For the last two decades, model-based emissions estimates are constrained by remote-sensing capacity to detect active fires and area burned. In AR5, biomass burning emissions were derived from a satellite product (Lamarque et al., 2010). Since then, improvements in detection of small fires has enhanced the agreement with higher-resolution and ground-based data on burned area in several regions (Randerson et al., 2012; Mangeon et al., 2015), especially for areas subjected to agricultural waste burning (Chuvieco et al., 2016, 2019). The updated emissions factors and the contribution of forest versus savanna fires lead to significantly higher global emissions of NOx and lower emissions of OC and CO in CMIP6, compared with CMIP5. A recent compilation and assessment of emissions factors (Andreae, 2019) indicates that the emissions factors from Akagi et al. (2011), primarily used to produce the CMIP6 datasets, differ by ±50% for CO, OC, BC and NOx, depending on the biome, and would imply, for example, up to 10–30% higher OC and BC emissions from tropical forest fires.
The historical (pre-satellite era) dataset for CMIP6 considers advances in knowledge of past fire dynamics (new fire proxy datasets, such as charcoal in sediments and levoglucosan in ice cores) and visibility records from weather stations (Marlon et al., 2016; van Marle et al., 2017). At a global level, CMIP5 and CMIP6 emissions trends are similar, however, there are substantial differences at the regional level, especially for the USA, South America (south of Amazonia) and Southern Hemisphere Africa (van Marle et al., 2017).
Globally, the CMIP5 estimates (Lamarque et al., 2010), indicated a gradual decline of open biomass burning emissions from 1920 to about 1950 and then steady, and stronger than CMIP6, increase towards 2000. In contrast, CMIP6 biomass burning emissions (van Marle et al., 2017) increase only slightly over 1750–2015 – they peak during the 1990s after which they decrease gradually, which is consistent with the assessment of fire trends in Chapter 5. Therefore, the CMIP6 evolution has a smaller difference between pre-industrial and present-day emissions than CMIP5, resulting in a lower radiative forcing of biomass burning SLCFs, possibly leading to a lower effect on climate (van Marle et al., 2017).
Climate warming, especially through change in temperature and precipitation, will generally increase the risk of fire (Jia et al., 2019, see also Chapter 12) and can also affect the fire injection and plume height (Veira et al., 2016), but occurrence of fires and their emissions in the future strongly depends on anthropogenic factors, such as population density, land use and fire management (Veira et al., 2016). Consequently, future emissions vary widely with increases and decreases amongst the SSP scenarios due to different land-use change scenarios.
In summary, there has been an improvement in the knowledge of biomass burning emissions by reducing key uncertainties highlighted in AR5. However, systematic assessment of remaining uncertainties is limited, with a lower limit of uncertainties due to emissions factors of 30%, and larger uncertainties due to burning-activity estimates, especially at regional level. Overall, a medium confidence in current global biomass burning SLCF emissions and their evolution over the satellite era is assessed. There is low to medium confidence in SLCF emissions from biomass burning from the pre-industrial period to the 1980s, which rely on the incorporation of several proxy data, with limited spatial representativeness. Nevertheless, uncertainties in the absolute value of pre-industrial emissions remain high, limiting confidence in radiative forcing estimates.
6.3 Evolution of Atmospheric SLCF Abundances
This section assesses the evolution of atmospheric abundance1 of SLCFs since AR5 based on observations and modelling, our knowledge of SLCF burden and distribution, and our understanding of the trends over longer time scales. In addition to emissions (Section 6.2), atmospheric chemistry (gas and heterogeneous chemistry), deposition (including wet and dry removal), and transport processes play a major role in determining the atmospheric distribution, budget and lifetime of SLCFs. The distribution and lifetime of SLCFs are further influenced by the modulation of chemical and physical processes in response to a changing climate. Therefore, the time evolution of atmospheric abundance of SLCFs is characterized by many complex non-linear interactions occurring at varying temporal and spatial scales. For this Assessment, global-scale, long-term measurements are employed only for a few gaseous SLCFs while for most short-lived species regional-scale observations and global models are relied upon.
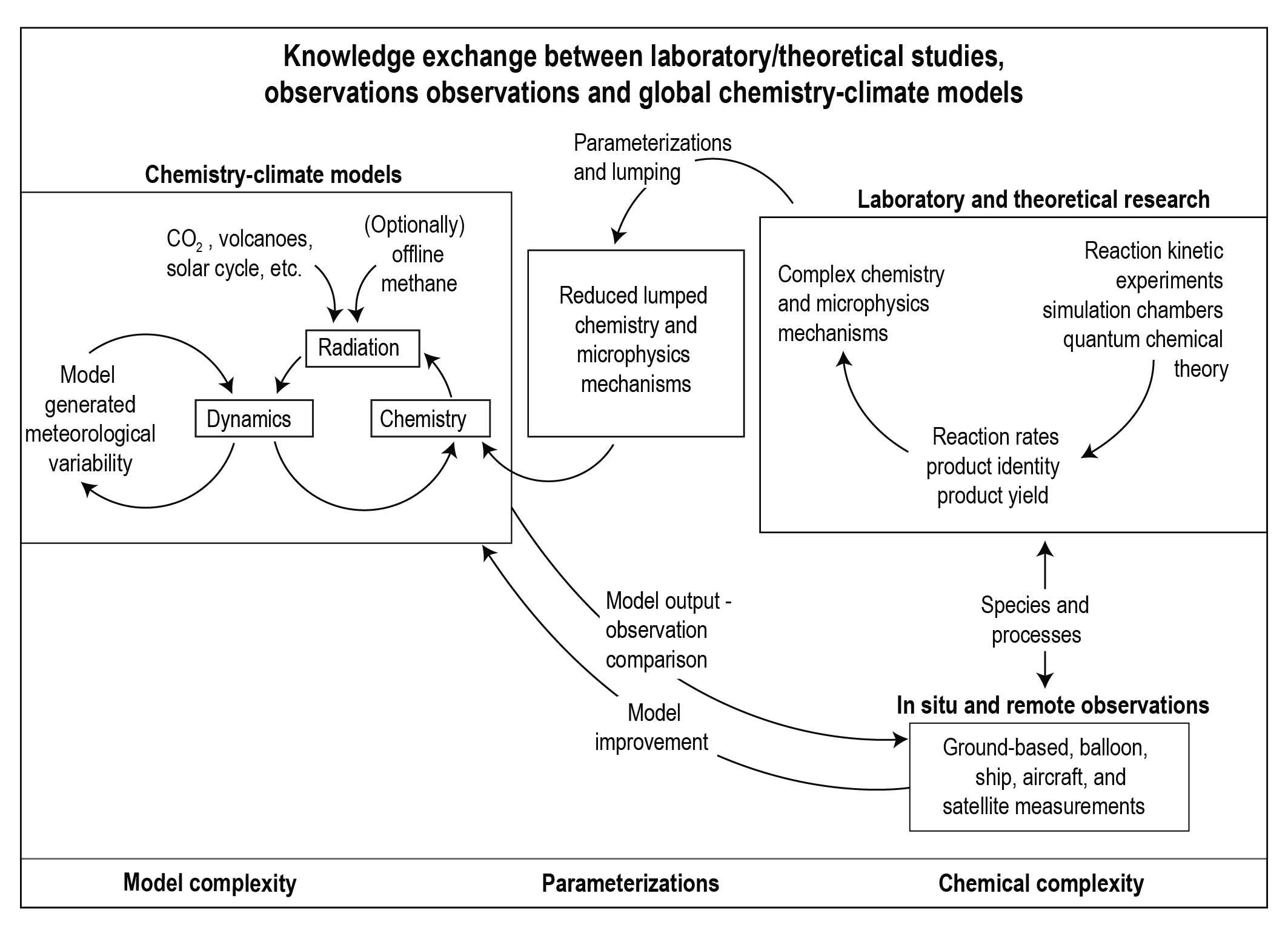
Box 6.1 | Atmospheric Abundance of SLCFs: From Process-level Studies to Global Chemistry–Climate Models
Changes in the atmospheric distribution of SLCFs determine their radiative forcing, and climate and air-quality impacts. This box provides an overview of how process-level understanding of the distribution and evolution of chemical compounds is derived and where uncertainties come from.
Process-level understanding of tropospheric gas and aerosol chemistry developed through laboratory and simulation chamber experiments, as well as quantum chemical theory, is used to generate chemical mechanisms. Atmospheric simulation chambers are designed to identify the chemical pathways and quantify reaction kinetics in isolation from atmospheric transport, deposition and emission processes. Ideally the chemical regimes studied are representative for ambient atmospheric complexity and concentrations (e.g., McFiggans et al. , 2019). Recently, quantum chemical theory has advanced to a level that it can provide kinetic and product information in a parameter range not possible with laboratory experiments (Vereecken et al. , 2015). Iterative and interlinked use of simulation chamber and quantum chemical theory has led to improved knowledge of chemical mechanisms (Peeters et al. , 2009, 2014; Nguyen et al. , 2010; Fuchs et al. , 2013). For application in chemistry–climate models (CCMs), the chemical mechanisms need to be computationally efficient, requiring simplifications. Such simplifications include reduced hydrocarbon representations, the application of lumping techniques (one compound or a chemical structure representing a family of compounds, for example, as done for parametrizing SOA formation) and/or the implementation of artificial operators representing key steps of the chemistry (Emmerson and Evans, 2009; Xia et al., 2009; Stockwell et al., 2020). Additionally, aerosol microphysical processes (nucleation, coagulation, condensation, evaporation and sedimentation) that determine the evolution of aerosol number concentrations and size particle distribution are represented in parametrized forms in global models with varying levels of complexity (Mann et al., 2014).
A wide range of in situ and remotely sensed observations are used to characterize atmospheric chemical composition. Measurements made routinely as part of long-term monitoring programmes are particularly useful for assessing long-term trends and variability, and spatial distributions (Sections 2.2, 6.3 and 7.3.3), while intensive field campaigns provide a more comprehensive view of atmospheric composition at a specific location for a limited time, facilitating an improved process-level understanding. Retrieval of atmospheric concentrations from satellites, in particular, has been tremendously useful for providing global continuous coverage, although the retrievals themselves depend on prior information of atmospheric composition usually derived from models. Over the last decade or so, observations of atmospheric concentrations have been combined with information from global chemistry–climate models to produce global assimilation and forecasting systems with the purpose of producing chemical reanalysis or improving model inputs (i.e., emissions or boundary conditions) and forecasts (Miyazaki et al., 2015; Randles et al., 2017; Inness et al., 2019).
Global three-dimensional CCMs (Box 6.1, Figure 1) represent the full coupling of chemistry with climate physics (e.g., Morgenstern et al. , 2017) with different levels of complexity (e.g., interactive aerosols with or without tropospheric and/or stratospheric chemistry). Methane concentrations are typically prescribed or constrained to observations while emissions of other SLCFs (or their precursors) are either prescribed or calculated interactively in the current generation of CCMs (Collins et al. , 2017). CCMs, now part of Earth system models (ESMs), are applied extensively to simulate the distribution and evolution of chemical compounds on a variety of spatial and temporal scales to improve current knowledge, make future projections and investigate global scale chemistry–climate interactions and feedbacks (Section 3.8.2.2). CCMs are also used to interpret observations to disentangle the processes that drive observed variability and trends. Some aspects of air quality, such as diurnal peaks or local threshold violations, strong gradient in chemical regimes and coupling between processes cannot be captured by relatively coarse spatial resolution (>50 km) global CCMs (Markakis et al., 2014) and necessitate subsequent downscaling modelling exercises.
The skill of CCMs is typically assessed by their ability to reproduce observed abundance, trends and variability of chemical compounds. However, uncertainty remains large because of observation limitations (errors and uncertainties, spatial and temporal coverage),
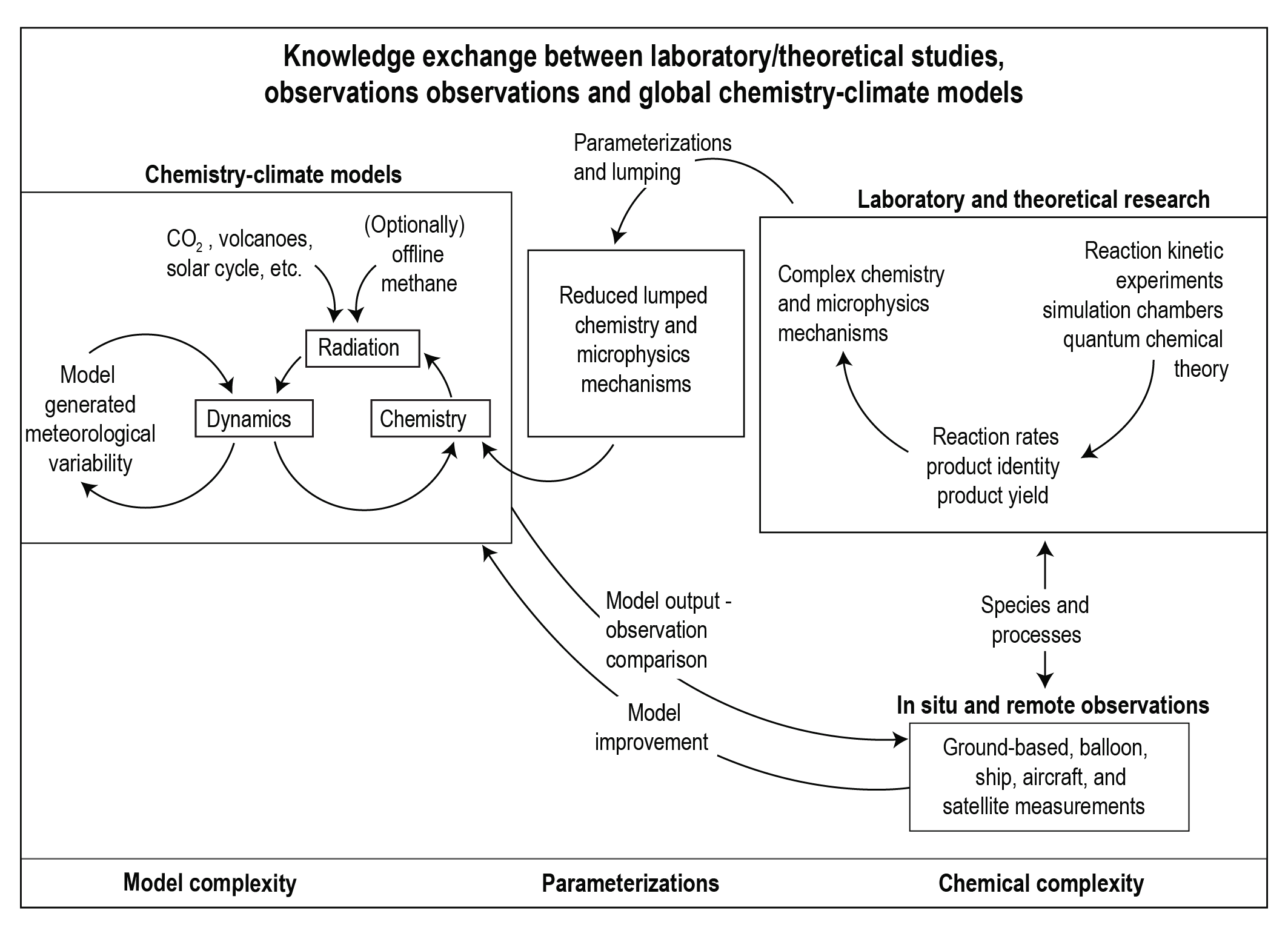
Box 6.1, Figure 1 | Knowledge exchange between laboratory/theoretical studies, observations and global climate–chemistry models (CCMs) to inform our understanding of short-lived climate forcers (SLCFs).
model parametrizations (e.g., chemical mechanisms, photolysis schemes, parametrizations for mixing and convective transport, and deposition), model input parameters (e.g., reaction rate constants, emissions) and an incomplete understanding of the physical and chemical processes that determine SLCF distributions (Brasseur and Jacob, 2017; Young et al., 2018). CCMs can therefore not capture every aspect of atmospheric chemical composition, but are expected to represent, as faithfully as possible, the sensitivity of chemical compounds to their drivers (e.g., anthropogenic emissions). Models are evaluated in multiple ways to identify their strengths and weaknesses in explaining the evolution of SLCF abundances. For example, CCM simulations are performed in the nudged or offline meteorology mode, that is, driven by observed or reanalysed meteorology rather than in the free-running mode, for consistent comparison of modelled chemical composition with observations for a specific time period (Dameris and Jöckel, 2013). However, caution is exercised as nudging can alter the model climate resulting in unintentional impacts on the simulated atmospheric physics and/or chemistry (Orbe et al., 2018; Chrysanthou et al., 2019). Chemical mechanisms implemented in CCMs are evaluated and intercompared to assess their skill in capturing relevant chemistry features (e.g., Brown-Steiner et al., 2018). The multi-model ensemble approach, employed for evaluating climate models, has been particularly useful for characterizing errors in CCM simulations of SLCFs related to structural uncertainty and internal variability (Naik et al. , 2013; Shindell et al. , 2013; Young et al. , 2013; Turnock et al. , 2020). However, as discussed in Box 4.1, this approach is unable to capture the full uncertainty range.
This assessment draws upon results from single-model studies and recent multi-model intercomparisons (e.g., AeroCom, CCMI), in particular those endorsed by CMIP6 (see Table 1.3), which then allows for the full consideration of robustness and uncertainty due to model structures and processes. Based on the collective information provided in this body of literature, the CMIP6 multi-model ensemble is largely fit-for-purpose of evaluating the influence of SLCFs on radiative forcing, climate and non-CO2 biogeochemical feedbacks. Additionally, CMIP6 models are fit for capturing the global air pollution response to changes in emissions and meteorology, but have difficulty in simulating the mean state (Turnock et al., 2020). The set of CMIP6 simulations has been used to update the relations between emissions and surface temperature at the heart of the emulators (Cross-Chapter Box 7.1) and update emissions metrics (Section 7.6). Emulators and emissions metrics are used in this chapter (Sections 6.6 and 6.7) to assess more specifically the effect of the individual SLCFs for each sector and region, which would be of prohibitive computing cost with CCMs. CCMs are also used to build global source-receptor models which use relations between surface concentrations and emissions. Such a model is used to assess the impact of various mitigation policies on air quality (Sections 6.5 and 6.7).
6.3.1 Methane (CH4)
The global mean surface mixing ratio of methane has increased by 156% since 1750 (Section 2.2.3.4 and Annex III). Since AR5, the methane mixing ratio has increased by about 3.5% from 1803 ± 2 ppb in 2011 to 1866 ± 3 ppb in 2019 (Section 2.2.3.3.2) largely driven by anthropogenic activities as assessed in Chapter 5 (Section 5.2.2 and Cross-Chapter Box 5.2).
An assessment of the global methane budget is provided in Chapter 5, while this section assesses methane atmospheric lifetime and perturbation time (Prather et al., 2001). The AR5 based its assessment of methane lifetime on Prather et al. (2012). The methane chemical lifetime due to tropospheric OH, the primary sink of methane, was assessed to be 11.2 ± 1.3 years constrained by surface observations of methyl chloroform (MCF), and lifetimes due to stratospheric loss, 2tropospheric halogen loss and soil uptake were assessed to be 150 ± 50 years, 200 ± 100 years, and 120 ± 24 years, respectively (Myhre et al., 2013). Considering the full range of individual lifetimes, the total methane lifetime was assessed in AR5 to be 9.25 ± 0.6 years.
The global chemical methane sink, essentially due to tropospheric OH, required to calculate the chemical lifetime is estimated by either bottom-up global CCMs and ESMs (BU) or top-down observational inversion methods (TD). BU global models represent the coupled chemical processes and feedbacks that determine the chemical sinks but show large diversity in their estimates, particularly the tropospheric OH sink (Zhao et al., 2019; Stevenson et al., 2020). TD inversion methods, on the contrary, provide independent observational constraints on the methane sink due to tropospheric OH over large spatio-temporal scales, but are prone to observational uncertainties and do not account for the chemical feedbacks on OH (Prather and Holmes, 2017; Naus et al., 2019). The central estimate of mean chemical methane loss over the period 2008–2017 varied from 602 [minimum and maximum range of 507–803] Tg yr–1 from BU chemistry–climate models in the Chemistry–Climate Modelling Initiative (CCMI) to 514 [474–529] Tg yr–1 from TD inverse modelling (Section 5.2.2 and Table 5.2). The smaller range in the TD estimate (11%) results from the use of a common climatological mean OH distribution (Saunois et al., 2020; Zhao et al., 2020a), while the larger range in the BU estimate (49%) reflects the diversity in OH concentrations from different chemical
mechanisms implemented in the global models (Zhao et al., 2019). See Section 6.3.6 for further discussion on the conflicting information on OH from CCMs/ESMs and TD inversion approaches. Further work is required to reconcile differences between BU and TD estimates of the chemical methane sink.
The present-day BU methane chemical lifetime shows a larger spread than that in the TD estimates (Table 6.2) in line with the spread seen in the sink estimates. The spread in the methane lifetime calculated by three CMIP6 ESMs is narrower and is enclosed within the spread of the BU CCMI model ensemble. Based on the consideration that the small imbalance in total methane sources versus sinks derived from TD estimates is close to the observed atmospheric methane growth rate (Table 5.2), the TD values are assessed to be the best estimates for this assessment. The relative uncertainty (± 1 standard deviation) is taken to be the same as that in AR5, that is, 11.8%, 33% and 10% for chemical, soil and total lifetime, respectively. The central estimate of the total atmospheric methane lifetime assessed here is the same as that in AR5.
Study | Total Chemical Lifetime (years) | Soil Lifetime (years) | Total Atmospheric Lifetime (years) | Number of Models/ Inversions |
8.3 (8.1–8.6)b | 160 | 8.0 (7.7–8.2) | 3 CMIP6 ESMs | |
Bottom-up (based on Table 5.2) | 8.3 (6.2–9.8) | 166 (102–453) | 8.0 (6.3–10.0) | 7 CCMI CCMs/CTMs |
Top-down (based on Table 5.2) | 9.7 (9.4–10.5) | 135 (116–185) | 9.1 (8.7–10.0) | 7 inversion systems |
AR6 assessed valuec | 9.7 ± 1.1 | 135 ± 44 | 9.1 ± 0.9 | Based on top-down with uncertainty estimate from AR5 |
aMean over 2005–2014
bDoes not include lifetime due to tropospheric halogen loss
cUncertainties indicate ±1 standard deviation
The methane perturbation lifetime (τpert ) is defined as the e-folding time it takes for the methane burden to decay back to its initial value after being perturbed by a change in methane emissions. Perturbation lifetime is longer than the total atmospheric lifetime of methane, as an increase in methane emissions decreases tropospheric OH, which in turn increases the lifetime and therefore the methane burden (Prather, 1994; Fuglestvedt et al., 1996; Holmes et al., 2013; Holmes, 2018). Since perturbation lifetime relates changes in emissions to changes in burden, it is used to determine the emissions metrics assessed in Chapter 7 (Section 7.6). The perturbation lifetime is related to the atmospheric lifetime asτpert = f *τtotal where f is the feedback factor and is calculated as f = 1/(1-s), where s = δ(lnτtotal)/ δ(ln[CH4]) (Prather et al., 2001). Since there are no observational constraints for eitherτpert or f, these quantities are derived from CCMs or ESMs. AR5 used f = 1.34 ± 0.06 based on a combination of multi-model (mostly CTMs and a few CCMs) estimates (Holmes et al., 2013). A recent model study explored new aspects of methane feedbacks finding that the strength of the feedback, typically treated as a constant, varies in space and time but will in all likelihood remain within 10% over the 21st century (Holmes, 2018). For this Assessment, the value of f is assessed to be 1.30 ± 0.07 based on a six-member ensemble of AerChemMIP ESMs (Thornhill et al., 2021b). This f value is slightly smaller but within the range of the AR5 value. This results in an overall perturbation methane lifetime of 11.8 ± 1.8 years, within the range of the AR5 value of 12.4 ± 1.4 years. The methane perturbation lifetime assessed here is used in the calculation of emissions metrics in Section 7.6.
6.3.2 Ozone (O3)
6.3.2.1 Tropospheric Ozone
About 10% of the total atmospheric ozone column resides in the troposphere. The ozone forcing on climate strongly depends on its vertical and latitudinal distribution in the troposphere. The lifetime of ozone in the troposphere ranges from a few hours in polluted urban regions to up to few months in the upper troposphere. Observed tropospheric ozone concentrations range from less than 10 ppb over the tropical Pacific Ocean to as much as 100 ppb in the upper troposphere and more than 100 ppb downwind of major ozone precursor emissions regions. An ensemble of five CMIP6 models including whole atmospheric chemistry and interactive ozone has been shown to simulate consistently the present-day ozone distribution (north to south and latitudinal gradients) and its seasonal variability when compared with observations from sondes, background surface stations and satellite products (Griffiths et al., 2021). The biases, whose magnitude is similar to AR5, are lower than 15% against climatological seasonal cycles from ozonesondes with an overestimate in the Northern Hemisphere and an underestimate in the Southern Hemisphere (Griffiths et al., 2021). The CMIP6 multi-model ensemble estimate of the global mean lifetime of ozone for present-day conditions is 25.5 ± 2.2 days (Griffiths et al., 2021), which is within the range of previous multi-model estimates (Stevenson et al., 2006; Young et al., 2013), indicating a high level of confidence.
The AR5 assessed the tropospheric ozone burden to be 337 ± 23 Tg for the year 2000 based on the ACCMIP ensemble of model simulations (Myhre et al., 2013). Multiple satellite products, ozonesondes and CCMs are used to estimate tropospheric ozone burden (Table 6.3). Satellite products provide lower-bound values as they exclude regions under polar night conditions (Gaudel et al., 2018). The tropospheric ozone burden values from multi-model exercises are within the range of the observational estimates despite different definitions of the tropopause for multi-model estimates which can lead to differences of about 10% on the ozone-burden model estimates (Griffiths et al., 2021). Weighted by their number of members, CMIP6 and CCMI multi-model estimates and observational estimates of tropospheric ozone burden in about the year 2010, lead to an assessment of the tropospheric ozone burden of 347 ± 28 Tg for 2010.
Period | Burden (Tg) | Production Tg yr–1 | Loss Tg yr–1 | Deposition Tg yr–1 | STE Tg yr–1 | Number of Models/Reference |
Models | ||||||
~2000 time slice (1995–2004) | 347 ± 30 | 4510 ± 566 | 3948 ± 379 | 846 ± 44 | 284 ± 193 | CMIP6a (5 Earth system models for burden and 4 models for budget terms) |
~2010 time slice (2005–2014) | 356 ± 31 | 4708 ± 589 | 4122 ± 399 | 863 ± 40 | 277 ± 201 | |
~2000 | 341 ± 31 (309 ± 31)* | CCMIb (9 models) (Archibald et al., 2020) | ||||
2010 | 345 ± 30 (314 ± 29)* | |||||
~2000 | 340 ± 34 | 4937 ± 656 | 4442 ± 570 | 996 ± 203 | 535 ± 161 | TOARc (based on 32–49 models participating in inter-model comparisons and single-model studies) |
Observations | ||||||
2010–2014 | 338 ± 6 | TOSTd, IASIe-FORLI, and IASI-SOFRID | ||||
2010–2014 | 302 ± 12* | TOST, IASI-FORLI, IASI-SOFRID, OMIf /MLS, OMI-SAO and OMI-RAL | ||||
aCMIP6: Coupled Model Intercomparison Project Phase 6;bCCMI: Chemistry–Climate Model Initiative;c toAR Tropospheric Ozone Assessment Report;d toST Trajectory-mapped Ozonesonde dataset for the Stratosphere and Troposphere;eIASI Infrared Atmospheric Sounding Interferometer;f OMI Ozone Monitoring Instrument.
The tropospheric ozone budget is controlled by chemical production and loss, by stratospheric–tropospheric exchange (STE), and by deposition at the Earth’s surface, whose magnitude are calculated by CCMs (Table 6.3). Despite The high agreement of the model ensemble mean with observational estimates in the present-day tropospheric ozone burden, the values of individual budget terms can vary widely across models in CMIP6, consistent with previous model intercomparison experiments (Young et al., 2018). Furthermore, single-model studies have shown that the halogen chemistry, which is typically neglected from model chemistry schemes in CCMs, may have a notable impact on the ozone budget, as halogens, particularly of marine origin, take part in efficient ozone-loss catalytic cycles in the troposphere (Saiz-Lopez et al. , 2012; Sarwar et al. , 2015; Sherwen et al. , 2016).
Because of the heterogeneous distribution of ozone, limited observations or proxies do not provide accurate information about the global pre-industrial abundance, posing a challenge to the estimation of the historical evolution of tropospheric ozone. Therefore, global CCMs complemented by observations are relied upon for estimating the long-term changes in tropospheric ozone. The AR5 concluded that anthropogenic changes in ozone precursor emissions are unequivocally responsible for the increase in tropospheric ozone between 1850 and the present (Myhre et al., 2013). Based on limited isotopic evidence, Chapter 2 assesses that the global tropospheric ozone increased by less than 40% between 1850 and 2005 (low confidence) (Section 2.2.5.3). The CMIP6 models are in line with this increase of tropospheric ozone with an ensemble-mean value of 109 ± 25 Tg (model range) from 1850–1859 to 2005–2014 (Figure 6.4). This increase is higher than the AR5 value of 100 ± 25 Tg from 1850–2010 due to higher ozone precursor emissions in CMIP6. However, the AR5 and CMIP6 values are close when considering the reported uncertainties. The uncertainties are equivalent in CMIP6 and AR5 despite enhanced inclusion of coupled processes in the CMIP6 ESMs (e.g., biogenic NMVOC emissions or interactive stratospheric ozone chemistry).
Since the mid-20th century, the CMIP6 model ensemble shows a higher global trend (Figure 6.4). Since the mid-1990s, the trends are better documented by observations (Section 2.2.5.3) and indicate spatial heterogeneity. In particular, in situ observations at remote surface sites and in the lower free troposphere indicate positive trends that are far more common than negative trends, especially in the northern tropics and across Southern and Eastern Asia (Figure 6.5). The CMIP6 ensemble and observations largely agree on the magnitude of the global positive trend since 1997 (0.82 ± 0.13 Tg yr–1 in the model ensemble; 0.70 ± 0.15 Tg yr–1 in the ozonesonde dataset; 0.83 ± 0.85 Tg yr–1 in the satellite ensemble) and qualitatively reproduce positive trends in the Southern Hemisphere (Griffiths et al., 2021). More analyses are needed for evaluation in other parts of the world to assess the skill of the recent ensemble based on CMIP6 emissions.
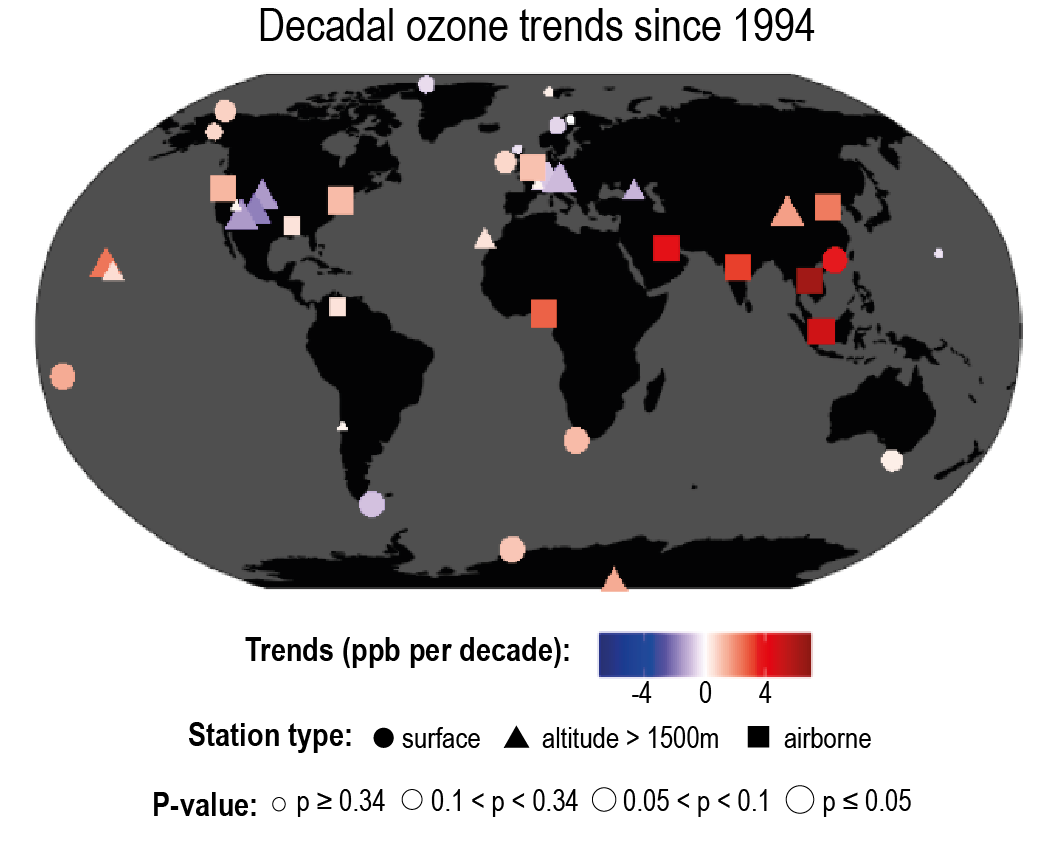 Figure 6.5 | Decadal tropospheric ozone trendssince 1994. Trends are shown at 28 remote and regionally representative surface sites (Cooper et al., 2020) and in 11 regions of the lower free troposphere (650 hPa, about 3.5 km) as measured by In-Service Aircraft for a Global Observing System (IAGOS) above Europe, north-eastern USA, south-eastern USA, western North America, north-east China, South East Asia, southern India, the Persian Gulf, Malaysia/Indonesia, the Gulf of Guinea and northern South America (Gaudel et al., 2020). High-elevation surface sites are >1500 m above sea level. All trends end with the most recently available year but begin in 1995 or 1994. The sites and datasets are the same as those used in Figure 2.8, further details on data sources and processing are available in the Chapter 2 data table (Table 2.SM.1).
Figure 6.5 | Decadal tropospheric ozone trendssince 1994. Trends are shown at 28 remote and regionally representative surface sites (Cooper et al., 2020) and in 11 regions of the lower free troposphere (650 hPa, about 3.5 km) as measured by In-Service Aircraft for a Global Observing System (IAGOS) above Europe, north-eastern USA, south-eastern USA, western North America, north-east China, South East Asia, southern India, the Persian Gulf, Malaysia/Indonesia, the Gulf of Guinea and northern South America (Gaudel et al., 2020). High-elevation surface sites are >1500 m above sea level. All trends end with the most recently available year but begin in 1995 or 1994. The sites and datasets are the same as those used in Figure 2.8, further details on data sources and processing are available in the Chapter 2 data table (Table 2.SM.1). In summary, there is high confidence in the estimated present-day (about 2010) global tropospheric ozone burden based on an ensemble of models and observational estimates (347 ± 28 Tg), but there is medium confidence among the individual models for their estimates of the tropospheric ozone-related budget terms.
Evidence from successive multi-model intercomparisons and the limited isotopic evidence agree on the magnitude of the increase of the tropospheric ozone burden from 1850 to the present day in response to anthropogenic changes in ozone precursor emissions corroborating AR5 findings. This increase is assessed to be 109 ± 25 Tg (medium confidence). The CMIP6 model ensemble shows a constant global increase since the mid-20th century whose rate is consistent with that derived from observations since the mid-1990s.
6.3.2.2 Stratospheric Ozone
Ninety percent of the total column ozone (TCO) resides in the stratosphere. The chemical lifetime of ozone in the stratosphere ranges from less than a day in the upper stratosphere to several months in the lower stratosphere (Bekki and Lefevre, 2009). Global stratospheric ozone trends based on observations are assessed in Chapter 2 (Section 2.2.5.2). The CMIP6 model ensemble shows that global TCO has slightly changed from 1850–1960 (Keeble et al., 2021). The rapid decline in the 1970s and 1980s due to halogenated ozone-depleting substances (ODSs, as assessed in Section 2.2.5.2 from observations) until the end of the 1990s, followed by a slight increase since then, is captured by the models (Keeble et al., 2021). Overall, the observed climatology patterns and annual cycle amplitudes are well represented in the CMIP6 ensemble mean. The CMIP6 ensemble overestimates the observed TCO values by up to 6% (10–20 Dobson Units (DU)) globally in the NH and SH mid-latitudes, and in the tropics, but the trend in these regions is well captured between 1960 and 2014. However, there is poor agreement between the individual CMIP6 models in the pre-industrial period and throughout the historical period, with model TCO values spread across a range of about 60 DU. The global stratospheric ozone column decreased by 14.3 ± 8.7 DU from 1850–2014 (Keeble et al., 2021).
Model simulations attribute about half of the observed upper-stratospheric ozone increase after 2000 to the decline of ODS since the late 1990s while the other half of the ozone increase is attributed to the slowing of gas-phase ozone destruction cycles due to cooling of the upper stratosphere by increasing GHGs (Aschmann et al., 2014; Oberländer-Hayn et al., 2015).
In summary, global stratospheric ozone column has decreased from pre-industrial period to present day in response to the ODS-induced ozone rapid decline in the 1970s and 1980s, followed by slow, and still incomplete, recovery. There is medium confidence that global stratospheric ozone column has changed by 14.3 ± 8.7 DU between 1850 and 2014.
6.3.3 Precursor Gases
6.3.3.1 Nitrogen Oxides (NOx)
The distribution of tropospheric NOx is highly variable in space and time owing to its short lifetime coupled with highly heterogeneous emission and sink patterns. NOx undergoes chemical processing, including the formation of nitric acid (HNO3), nitrate (NO–3), and organic nitrates (e.g., alkyl nitrate and peroxyacyl nitrate), atmospheric transport, and deposition. Despite challenges in retrieving quantitative information from satellite observations (Duncan et al., 2014; Lin et al., 2015; Lorente et al., 2017; Silvern et al., 2018), improved accuracy and resolution of satellite-derived tropospheric NO2 columns over the past two decades have advanced understanding of the global distribution, long-term trends and source attribution of NOx. Long-term average tropospheric NO2 column based on multiple satellite-borne instruments (Figure 6.6a) reveals the highest NO2 levels over the most populated, urbanized and industrialized regions of the world corresponding to high NOx emissions source regions (Krotkov et al., 2016; Georgoulias et al., 2019). Enhanced but highly variable NO2 columns are also associated with biomass-burning regions as well as areas influenced by lightning activity (Miyazaki et al., 2014; Tanimoto et al., 2015).
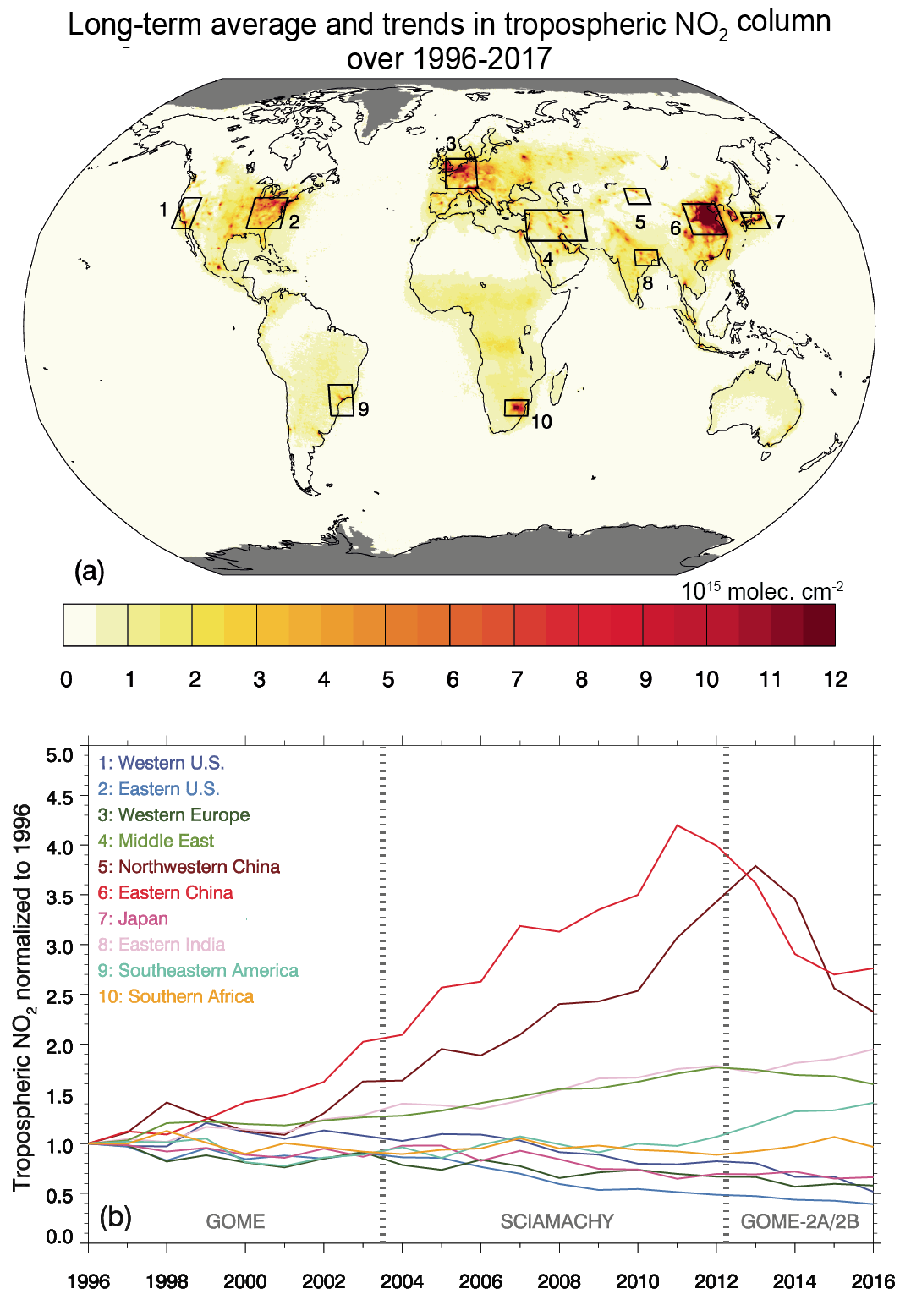 Figure 6.6 | Long-term climatological mean (a) and time evolution (b) of tropospheric nitrogen dioxide (NO2 ) vertical column density. Data are from the merged GOME/SCIAMACHY/GOME-2 (TM4NO2A version 2.3) dataset for the period 1996–2016 (Georgoulias et al., 2019). Time evolution of NO2 column shown in panel (b) is normalized to the fitted 1996 levels for the 10 regions shown as boxes in panel (a). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.6 | Long-term climatological mean (a) and time evolution (b) of tropospheric nitrogen dioxide (NO2 ) vertical column density. Data are from the merged GOME/SCIAMACHY/GOME-2 (TM4NO2A version 2.3) dataset for the period 1996–2016 (Georgoulias et al., 2019). Time evolution of NO2 column shown in panel (b) is normalized to the fitted 1996 levels for the 10 regions shown as boxes in panel (a). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). Observational constraints derived from the isotopic composition of atmospheric nitrate inferred from ice cores provide evidence of increasing anthropogenic NOx sources since pre-industrial times (Hastings et al., 2009; Geng et al., 2014). Global NOx emissions trends in bottom-up inventories (Section 6.2.1) as well as model simulations of nitrogen deposition (Lamarque et al., 2013a) are in qualitative agreement with these observational constraints. CMIP6 ESMs exhibit stable NOx4 burden over the first half of the 20th century and then a sharp increase driven by a factor of three increase in emissions, however, the magnitude of this increase remains uncertain due to poor observational constraints on pre-industrial concentrations of NOx (Griffiths et al., 2021).
The AR5 reported NO2 decreases by 30–50% in Europe and North America, and increases by more than a factor of two in Asia, over the 1996–2011 period based on satellite observations (Hartmann et al., 2013). Extension of this analysis covering the time period up to 2015 reveals that NO2 has continued to decline over the USA, Western Europe and Japan (Schneider et al., 2015; Duncan et al., 2016; Krotkov et al., 2016) because of effective fossil fuel NOx emissions controls (Section 6.2), although this rate of decline has slowed down post-2011 (Jiang et al., 2018). Satellite observations also reveal a 32% decline in NO2 column over China after peaking in 2011 (Figure 6.6b), consistent with declining NOx emissions (Section 6.2) due to the implementation of emissions-control strategies (de Foy et al. , 2016; Irie et al. , 2016; F. Liu et al. , 2016). Over Southern Asia, tropospheric NO2 levels have grown rapidly with increases of 50% during 2005–2015, largely driven by hotspot areas in India experiencing rapid expansion of the power sector (Duncan et al., 2016; Krotkov et al., 2016). Further analysis indicates that many parts of India have also undergone a reversal in NO2 trends since 2011 that has been attributed to a combination of factors, including a slowdown in economic growth, implementation of cleaner technologies, non-linear NOx chemistry, and meteorological variability (Georgoulias et al., 2019). Satellite data reveals spatially heterogeneous NO2 trends over the Middle East with an overall increase over 2005–2010 and a decrease over large parts of the region after 2011–2012. The reasons for trend reversal within individual areas are diverse, including warfare, imposed sanctions, and air-quality controls (Lelieveld et al., 2015a; Georgoulias et al., 2019). Satellite-derived tropospheric NO2 levels over Africa and Latin America do not show a clear trend; both increasing and decreasing trends are observed over large agglomerations in these regions since the early 2000s (Schneider et al., 2015; Duncan et al., 2016).
In summary, global tropospheric NOx abundance has increased from 1850–2015 (high confidence). Satellite observations of tropospheric NOx indicate strong regional variations in trends over 2005–2015. There is high confidence that NO2 has declined over the USA and Western Europe since the mid-1990s and increased over China until 2011. NO2 trends have reversed (declining) over China beginning in 2012 and NO2 has increased over Southern Asia by 50% since 2005 (medium confidence).
Analysis Period | Trends: Regions | Reference/Methodology |
Global/Hemispheric | ||
2003–2015 | –0.86% yr–1 | Flemming et al. (2017)Model assimilating MOPITT |
2002–2013 | –1.4% yr–1 | Model assimilating MOPITT |
2002–2018 | –0.50 ± 0.3% yr–1: 60°N–60°S (MOPITT) –0.56 ± 0.3% yr–1;–0.61 ± 0.2% yr–1: 0°–60°N –0.35 ± –0.3% yr–1; -0.33±0.3% yr–1: 0°–60°S | Satellite Observations MOPITT; AIRS |
2000–2017 | –0.32 ± 0.05% yr–1 | Satellite Observations MOPITT |
2003–2014 | around –2.5 to 0.5 ppb yr–1: Northern Hemisphere around –0.5 to 0 ppb yr–1: Southern Hemisphere | Flemming et al. (2017) NOAA Carbon Cycle Cooperative Global Air Sampling Network |
2001–2013 | –2.19 to –0.80 ppb yr–1: Northern Hemisphere (Upper Troposphere/Tropopause Layer) | Cohen et al. (2018)IAGOS Airborne |
Pacific/Tropics | ||
2004–2013 (Spring Mean) | –2.9 ± 2.6 ppb yr–1: Mauna Loa (19.54°N, 155.58°W) | Gratz et al. (2015)Ground-based |
2004–2013 (Spring Mean) | –2.6 ± 1.8 ppb yr–1: Sand Island Midway (28.21°N, 177.38°W) | Gratz et al. (2015)Ground-based |
Europe | ||
1996–2006 | –0.45 ± 0.16% yr–1: Jungfraujoch (46.6°N, 8.0°E) –1.00 ± 0.24% yr–1: Zugspitze (47.4°N, 11.0°E) –0.62 ± 0.19% yr–1: Harestua (60.2°N, 10.8°E) 0.61 ± 0.16% yr–1: Kiruna (67.8°N, 20.4°E) | Angelbratt et al. (2011)Ground-based |
2001–2011 May to Sep | –3.1 ± 0.30 ppb yr–1: Pico Mt. Obs (38.47°N, 28.40°W) –1.4 ± 0.20 ppb yr–1: Mace Head, Ireland | Kumar et al. (2013)Ground-based |
2002–2018 | –-0.89 ± 0.1% yr–1: Europe (45°N–55°N, 0°E–15°E) | Satellite Observations MOPITT |
North America | ||
2001–2010 | –2.5 ppb yr–1: Thompson Farm (43.11°N, 70.95°W) –2.3 ppb yr–1: Mt. Washington (44.27°N, 71.30°W) +2.8 ppb yr–1: Castle Springs (43.75°N, 71.35°W) –3.5 ppb yr–1: Pack Monadnock (42.86°N, 71.88°W) –2.8 ppb yr–1: Whiteface Mountain (44.40°N, 73.90°W) –4.3 ppb yr–1: Pinnacle State Park (42.09°N, 77.21°W) | Zhou et al. (2017)Ground-based |
2004–2013 (Spring Mean) | –3.2 ± 2.9 ppb yr–1: Mt. Bachelor Observatory | Gratz et al. (2015)Ground-based |
2004–2012 (Spring Mean) | –2.8 ± 1.8 ppb yr–1: Shemya Island (55.21°N, 162.72°W) | Gratz et al. (2015)Ground-based |
2002–2018 | –0.85 ± 0.1%yr–1: Eastern USA (35°N–40°N, –95°E–75°E) | Satellite Observations MOPITT |
Asia | ||
2005–2018 | –0.46 ± 0.14% yr–1: Eastern Asia | WDCGG Ground-based |
2005–2018 | –0.41 ± 0.09% yr–1: Eastern Asia | MOPITT |
2002–2018 | –1.18 ± 0.3% yr–1: (Northeast China 30°E–40°E, 110°E–123°E) –0.28 ± 0.2% yr–1: (North India 20°N–30°N, 70°E–95°E) | Satellite Observations MOPITT |
6.3.3.2 Carbon Monoxide (CO)
About half of the atmospheric CO burden is due to its direct emissions and the remainder is due to the atmospheric oxidation of methane and NMVOCs. Reaction with OH is the primary sink of CO with a smaller contribution from dry deposition.
Since AR5, advances in satellite retrievals (e.g., Worden et al. , 2013; Warner et al. , 2014; Buchholz et al. , 2021), ground-based column observations (e.g., Zeng et al. , 2012; Té et al. , 2016), airborne platforms (e.g., Cohen et al., 2018; Petetin et al., 2018), surface measurement networks (e.g., Andrews et al. , 2014; Schultz et al. , 2015; Prinn et al. , 2018; Pétron et al. , 2019) and assimilation products (e.g., Deeter et al., 2017; Flemming et al., 2017; Zheng et al., 2019) have resulted in better characterization of the present-day atmospheric CO distribution. Typical annual mean surface CO concentrations range from around 120 ppb in the Northern Hemisphere to around 40 ppb in the Southern Hemisphere (Pétron et al., 2019). The sub-regional patterns in CO reflect the distribution of emissions sources. Seasonal hotspots are linked to areas of biomass burning in tropical South America, equatorial Africa, South East Asia and Australia. A study using data assimilation techniques estimates a global mean CO burden of 356 ± 27 Tg over the 2002–2013 period (Gaubert et al., 2017).
Global models generally capture the global spatial distribution of the observed CO concentrations but have regional biases of up to 50% (e.g., Emmons et al., 2020; Horowitz et al., 2020). Despite updated emissions datasets, the global multi-model and single-model simulations persistently underestimate observed CO concentrations at northern high and mid-latitudes as well as in the Southern Hemisphere, but with smaller biases compared with that in the Northern Hemisphere (Naik et al. , 2013; Stein et al. , 2014; Monks et al. , 2015; Strode et al. , 2015). Models are biased high in the tropics, particularly over highly polluted areas in India and Eastern Asia (Strode et al., 2016; Yarragunta et al., 2017).
Estimates of global CO burden simulated by global models generally fall within the range of that derived from data assimilation techniques, though the spread across the models is large (Naik et al. , 2013; Stein et al. , 2014; Zeng et al. , 2015; Myriokefalitakis et al. , 2016). There is a large diversity in model-simulated CO budget driven by uncertainties in CO sources and sinks, particularly those related to in situ production from NMVOCs and loss due to reaction with OH (Stein et al., 2014; Zeng et al., 2015; Myriokefalitakis et al., 2016). Global CO budget analysis from a multi-model ensemble for more recent years, including results from the CMIP6 model runs, are not yet available.
Reconstructions of CO concentrations based on limited ice-core samples in the Northern Hemisphere high latitudes suggest CO mole fractions of about 145 ppb in the 1950s, which rose by 10–15 ppb in the mid- 1970s, and then declined by about 30–130 ppb by 2008 (Petrenko et al., 2013). The negative trends since the 1990s are often attributed to emissions regulations from road transportation in North America and Europe. Due to limited observations prior to the satellite era, long-term global CO trends are based on estimates from models. An increase of global CO burden of about 50% for the year 2000 relative to 1850 is found in CMIP6 (Griffiths et al., 2021).
The AR5 reported a global CO decline of about 1% yr–1based on satellite data from 2002–2010, but biases in instruments rendered low confidence in this trend. The AR5 also indicated a small CO decrease from in situ networks but did not provide quantitative estimates. New analysis of CO trends performed since AR5 and based on different observational platforms and assimilation products show a decline globally and over most regions during the last one to two decades with varying amplitudes partly depending on the period of analysis (Table 6.4). Inversion-based analysis attributes the global CO decline during the past two decades to decreases in anthropogenic and biomass-burning CO emissions despite probable increase in atmospheric CO chemical production (Gaubert et al. , 2017; Jiang et al. , 2017; Zheng et al. , 2019). Furthermore, Buchholz et al. (2021) report a slowdown in global CO decline in 2010–2018 compared to 2002–2010, although the magnitude and sign of this change in the trend varies regionally. Global models prescribed with emissions inventories developed prior to the CMIP6 inventory capture the declining observed CO trends over North America and Europe but not over Eastern Asia (Strode et al., 2016). CMIP6 models driven by CMIP6 emissions simulate a negative trend in global CO burden over the 1990–2020 period (Griffiths et al., 2021), however the simulated trends have not yet been evaluated against observations.
In summary, our understanding of present-day global CO distribution has increased since AR5 with newer and improved observations and reanalysis. There is high confidence that global CO burden is declining since 2000. Evidence from observational CO reanalysis suggests this decline is driven by reductions in anthropogenic CO emissions, however this is yet to be corroborated by global ESM studies with the most recent emissions inventories.
6.3.3.3 Non-Methane Volatile Organic Compounds (NMVOCs)
NMVOCs encompass thousands of compounds with lifetimes from hours to days to months, and abundances and chemical composition highly variable with respect to space and time. Although the biogenic source (Section 6.2.2) dominates the global NMVOC budget, anthropogenic activities are the main driver of long-term trends in the abundance of many compounds.
Information on the global distribution of individual NMVOCs is scarce, except for the less reactive compounds having lifetimes of several days to months. Based on measurements from polar firn air samples and ground-based networks, AR5 reported that the abundances of the predominantly anthropogenic light alkanes (C2-C5) increased until 1980 and declined afterwards. The decline was attributed to air-quality emissions controls and to fugitive emissions decreases following the collapse of the Soviet Union (Simpson et al., 2012). Since AR5, scarce ground-based measurements have shown that the decline in C2-C3 alkanes ended around 2008 and their abundances are since growing again, which is primarily attributed to increasing North American emissions (Section 6.2.1). Furthermore, since AR5 the evolution of ethane levels during the past millennium was made accessible by analysis of ice-core samples (Nicewonger et al., 2016). The large observed interpolar ratio of ethane in pre-industrial times (3.9) corroborates a large geologic source of ethane previously put forward by (Etiope and Ciccioli, 2009), and narrows down its likely global magnitude (Nicewonger et al., 2018) (low to medium confidence). The incorporation of geologic emissions in CCMs is not yet systematic though a one-model study has shown improved agreement of the results with observations (Dalsøren et al., 2018).
Formaldehyde (HCHO) is a short-lived, high-yield product of NMVOC oxidation, and formaldehyde column data from satellite instruments can therefore inform on trends in anthropogenic NMVOC abundances over very industrialized regions. The AR5 reported significant positive trends in formaldehyde between 1997 and 2009 over northeastern China (4% yr–1) and negative trends over northeastern US cities. Since AR5, there is robust evidence and high agreement of an upward trend of HCHO over eastern China, though large regional disparities exist in the trends (De Smedt et al., 2015; Shen et al., 2019) with a possible negligible or decreasing trend over Beijing and the Pearl River Delta. In other world regions, in particular North America, there is limited to medium evidence for significant changes in the HCHO columns, except in regions where the trend is particularly strong (e.g., the Houston area: –2.2% yr–1 over 2005–2014) and the Alberta oil sands (+3.8% yr–1;Zhu et al., 2017). Over the northeastern USA, even the sign of the trend differs between studies (De Smedt et al. , 2015; Zhu et al. , 2017) for reasons that are unclear.
In summary, after a decline between 1980 and 2008, abundances of light NMVOCs have increased again over the Northern Hemisphere due to the extraction of oil and gas in North America (high confidence). Trends in satellite HCHO observations, used as a proxy of anthropogenic NMVOC over industrialized areas, show a significant positive trend over eastern China (high confidence) but also indicate large regional disparities in the magnitude of the trends over China and even in their signs over North America.
6.3.3.4 Ammonia (NH3)
Ammonia is the most abundant alkaline gas in the atmosphere. Its present-day source is dominated by livestock and crop production (Section 6.2). Ammonia reacts with nitric acid and sulphuric acid to produce ammonium sulphate and ammonium nitrate, which contribute to the aerosol burden (Section 6.3.5.2), promotes aerosol nucleation by stabilizing sulphuric acid clusters (Kirkby et al., 2011), and contributes to nitrogen deposition (Section 6.4.4; Sheppard et al., 2011; Flechard et al., 2020). Trends in NH3 were not assessed in AR5.
Considerable expansion of satellite (Clarisse et al. , 2009; Shephard and Cady-Pereira, 2015; Warner et al. , 2016) and ground-based observations (Miller et al. , 2014; Y. Li et al. , 2016; Pan et al. , 2018) has improved our understanding of the spatial distribution and seasonal to interannual variability of ammonia, and advanced its representations in models (e.g., Zhu et al., 2015). Regionally, peak NH3 concentrations are observed over large agricultural (e.g., northern India, the USA Midwest and Central Valley) and biomass-burning regions, in good qualitative agreement with emissions inventories (Van Damme et al. , 2015, 2018). However, several large agricultural and industrial hotspots have been found to be missing or greatly underestimated in emissions inventories (Van Damme et al. , 2018). NH3 exhibits a strong vertical gradient, with a maximum in the boundary layer (Schiferl et al., 2016), and can be transported into the upper troposphere and lower stratosphere (UTLS), particularly in the Asian Monsoon region, as indicated by observations (Froyd et al., 2009; Höpfner et al., 2016, 2019) and theoretical considerations (Ge et al. , 2018). There is a large range in the present-day NH34 burden (from 0.04–0.7 TgN) simulated by CCMs, highlighting deficiencies in the process-level representation of NH3 in current global models (Bian et al., 2017). The underestimate of surface NH3 concentrations (Bian et al., 2017) further highlights such deficiencies and the limitations in comparing site-specific observations with relatively coarse-resolution models.
Observations show that NH3 concentration has been increasing in recent decades in the USA (Butler et al. , 2016; Warner et al. , 2016; Yu et al. , 2018), Western Europe (van Zanten et al. , 2017; Warner et al. , 2017; Wichink Kruit et al. , 2017; Tang et al. , 2018), and China (Warner et al., 2017; M. Liu et al., 2018). This trend has been attributed to a combination of increasing ammonia emissions (Sutton et al., 2013; Fowler et al., 2015) and decreases in the chemical reaction of NH3 with nitric and sulphuric acids associated with reductions in SO2 and NOx emissions whose rate depends on the region (Warner et al., 2017; Yao and Zhang, 2019). Over longer time scales, CCMs simulate an increase of the NH34 burden by a factor of two to seven since pre-industrial conditions (Xu and Penner, 2012; Hauglustaine et al., 2014).
In summary, progress has been made in the understanding of the spatio-temporal distribution of ammonia, though representation of NH3 remains rather unsatisfactory due to process-level uncertainties. Evidence from observations and models suggests that ammonia concentrations have been increasing over recent decades due to emissions and chemistry. There is high confidence that the global NH34 burden has increased considerably from the pre-industrial period to the present day, although the magnitude of the increase remains uncertain.
6.3.3.5 Sulphur Dioxide (SO2)
The AR5 did not assess trends in SO2 concentrations. Trends in SO2 abundances are consistent with the overall anthropogenic emissions changes as presented in Section 6.2 and Figure 6.18. Long-term surface-based in situ observations in North America and Europe show reductions of more than 80% since the measurements began around 1980 (Table 6.5). Europe had the largest reductions in the first part of the period while the highest reduction came later in North America. Observed trends are qualitatively reproduced by global and regional models over North America and Europe during the period 1990–2015 for which emissions changes are well quantified (Table 6.5; Aas et al., 2019).
Analysis Period | Trends in SO2 | Trends in Particulate SO42– | Reference |
Global Models/Assimilated Models | |||
1990–2000 | –8.54 ± 1.40% yr–1 (EU, 43 sites) –2.63 ± 0.30% yr–1 (NA, 53 sites) | –5.23 ± 1.17% yr–1 (EU, 41 sites) –1.94 ± 0.43% yr— (NA 101 sites) | |
2000–2015 | –0.41 ± 0.92% yr–1 (EA, 19 sites) –4.86 ± 1.31% yr–1 (EU, 47 sites) –4.40 ± 0.93% yr–1 (NA, 77 sites) | 0.02 ± 0.91% yr–1 (EA, 13 sites) –3.26 ± 0.85% yr–1 (EU, 36 sites) –3.18 ± 0.66% yr–1 (NA, 218 sites) | |
Ground-based In Situ Observations | |||
1980–1990 | –5.03 ± 2.04% yr–1 (EU, 20 sites) –2.5% yr–1 (US) | –2.56 ± 3.10% yr–1 (EU, 16 sites) –1.80 ± 4.09% yr–1 (US SO42– in precipitation, 78 sites) | |
1990–2000 | –7.56 ± 1.81% yr–1 (EU, 43 sites) –3.27 ± 1.69% yr–1 (NA, 53 sites) | –5.16 ± 2.11% yr–1 (EU, 41 sites) –2.08 ± 1.44% yr–1 (NA, 101 sites) | |
2000–2015 | –0.14 ± 5.32% yr–1 (EA, 19 sites) –3.89 ± 2.16% yr–1 (EU, 47 sites) –4.69 ± 1.35% yr–1 (NA, 77 sites) | 2.68 ± 9.41% yr–1 (EA, 13 sites) –2.67 ± 2.03% yr–1 (EU, 36 sites) –3.15 ± 1.30% yr–1 (NA, 218 sites) | |
Change Based on Satellite Observations | |||
2005–2015 | ca –80% (Eastern US) | ||
2005–2015 | ca –60% (Eastern EU) | ||
2005–2015 | 200 ± 50% (India) | ||
2005 (and 2012) –2015 | ca –50% (The North China Plain) | ||
In situ observations over other parts of the world are scattered. However, the limited in situ observations in Eastern Asia indicate an increase in atmospheric SO2 up to around 2005 and then a decline (Aas et al., 2019). This is confirmed by satellite observations (Krotkov et al., 2016), which further reveal a rapid decline in SO2 since around 2012 or 2013 (Krotkov et al., 2016; Zheng et al., 2018b). In India, on the other hand, SO2 levels have doubled between 2005 and 2015 (Krotkov et al., 2016).
In summary, surface and satellite observations indicate strong regional variations in trends of atmospheric SO2 abundance. The SO2 concentrations in North America and Europe have declined over 1980–2015 with slightly stronger reductions in North America (70 ± 20%) than in Europe (58 ± 32%) over 2000–2015, though Europe had larger reductions than the US in the prior decade (1990–2000). In Asia, the SO2 trends are more scattered, though there is medium confidence that there was a strong increase up to around 2005, followed by a steep decline in China, while over India, the concentrations are increasing steadily.
6.3.4 Short-lived Halogenated Species
Halogenated species are emitted in the atmosphere in the form of the synthetically produced chlorofluorocarbons (CFCs), halons, hydrochlorofluorocarbons (HCFCs), hydrofluorocarbons (HFCs) and others. Their historical global abundances are provided in Annex III and discussed in Chapter 2 (Section 2.2.4 and Table 2.3). In summary, for the period 2011–2019, the abundance of total chlorine from HCFCs has continued to increase in the atmosphere with decreased growth rates; total tropospheric bromine from halons and methyl bromide continued to decrease while abundances of most currently measured HFCs increased significantly, consistent with expectations based on the ongoing transition away from the use of ODSs. Here, emphasis is given on the very short-lived halogenated species (VSLSs). The trends for these species were not discussed in IPCC AR5.
VSLSs are halogenated substances with atmospheric lifetimes less than half a year. While longer-lived ODSs account for most of the present-day stratospheric halogen loading, there is robust evidence that VSLSs contribute to stratospheric bromine and chlorine (Carpenter et al. , 2014; Elvidge et al. , 2015a; Hossaini et al. , 2015), thus also contributing to stratospheric ozone depletion.
Of the atmospheric VSLSs, brominated and iodinated species are predominantly of oceanic origin, while chlorinated species have significant additional anthropogenic sources (Carpenter et al., 2014; Hossaini et al., 2015). Global mean chlorine from the VSLSs has increased in the troposphere from about 91 ppt in 2012 to about 110 ppt in 2016 (Engel et al., 2018). This increase is mostly due to dichloromethane (CH2 cl2), a species that has predominantly anthropogenic sources reflected by three-times higher concentrations in the Northern Hemisphere than in the Southern Hemisphere (Hossaini et al., 2017). The upward dichloromethane trend is corroborated by upper-tropospheric aircraft data over the period 1998–2014 (Elvidge et al., 2015b; Oram et al., 2017). The observations from the surface networks show that the abundance of dichloromethane continued to increase until 2019 (Annex III), although the accuracy of global abundance of VSLSs is limited by the scarce coverage by networks. No long-term changes of the bromine-containing VSLSs have been observed (Engel et al., 2018).
6.3.5 Aerosols
This section assesses trends in the atmospheric distribution of aerosols and improvements in relevant physical and chemical processes. The observed large-scale temporal evolution of aerosols is assessed in Section 2.2.6. Since AR5, long-term measurements of aerosol mass concentrations from regional global surface networks have continued to expand and provide information on the distribution and trends in aerosols (Figure 6.7). There is large spatial variability in aerosol mass concentration, expressed as PM2.5, dominant aerosol type and aerosol composition, consistent with the findings in AR5.
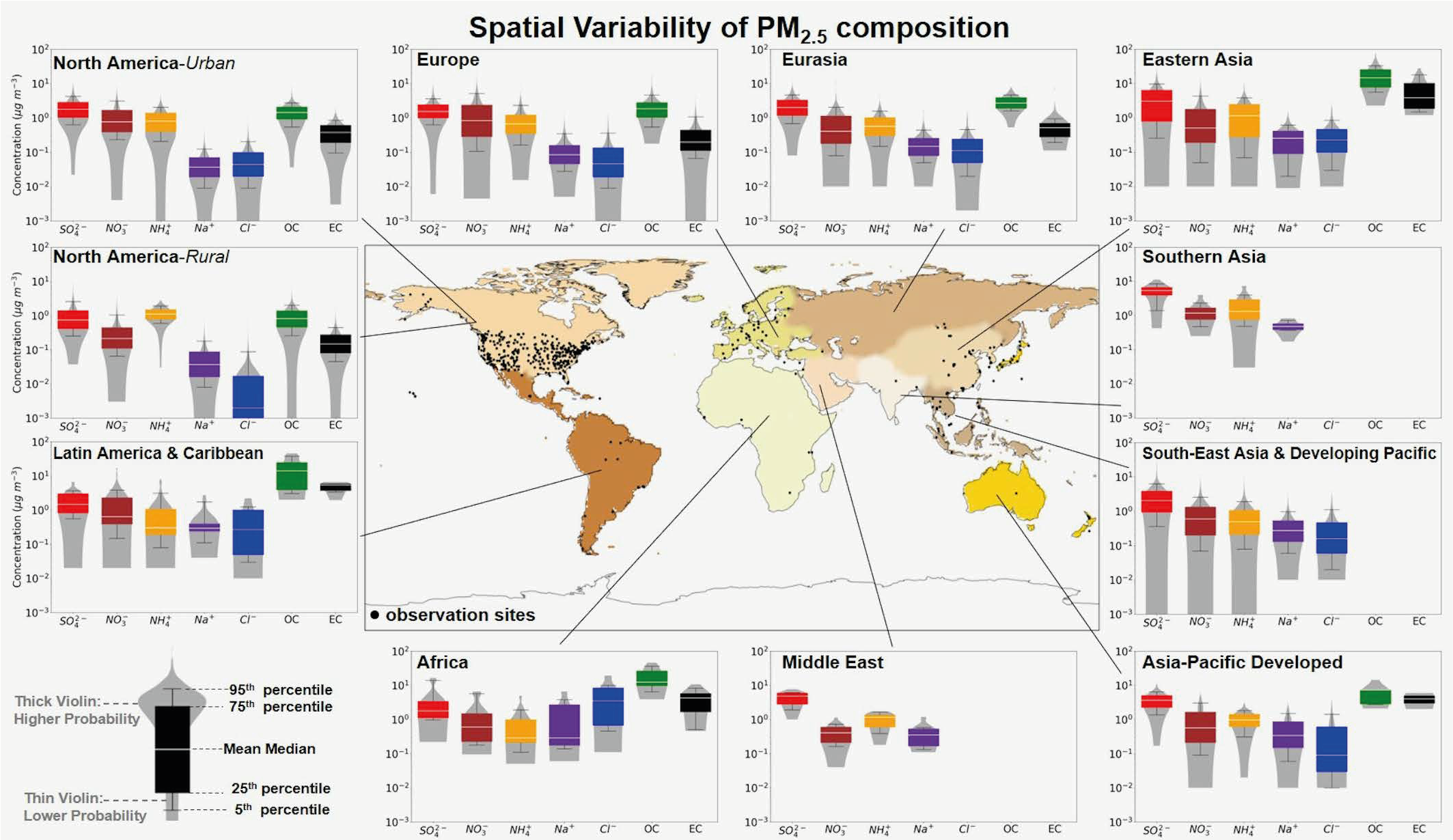 Figure 6.7 | Distribution of PM2.5composition mass concentration (in μg m–3) for the major PM2.5aerosol components. Those aerosol components are sulphate, nitrate, ammonium, sodium, chloride, organic carbon and elemental carbon. The central world map depicts the intermediate-level regional breakdown of observations (10 regions) following the IPCC Sixth Assessment Report Working Group III (AR6 WGIII). Monthly averaged PM2.5 aerosol component measurements are from: (i) the Environmental Protection Agency (EPA) network which include 211 monitor sites primarily in urban areas of North America during 2000–2018 (Solomon et al., 2014), (ii) the Interagency Monitoring of Protected Visual Environments (IMPROVE) network during 2000–2018 over 198 monitoring sites representative of the regional haze conditions over North America, (iii) the European Monitoring and Evaluation Programme (EMEP) network over 70 monitoring in Europe and (eastern) Eurasia during 2000–2018, (iv) the Acid Deposition Monitoring Network in Eastern Asia (EANET) network with 39 (18 remote, 10 rural, 11 urban) sites in Eurasia, Eastern Asia, South East Asia and Developing Pacific, and Asia-Pacific Developed during 2001–2017, (v) the global Surface Particulate Matter Network (SPARTAN) during 2013–2019 with sites primarily in highly populated regions around the world (i.e., North America, Latin America and Caribbean, Africa, Middle East, Southern Asia, Eastern Asia, South East Asia and Developing Pacific; Snider et al., 2015, 2016), and (vii) individual observational field campaign averages over Latin America and Caribbean, Africa, Europe, Eastern Asia, and Asia-Pacific Developed (Celis et al., 2004; Feng et al., 2006; Bourotte et al., 2007; Fuzzi et al., 2007; Mariani and de Mello, 2007; Molina et al., 2007, 2010; Favez et al., 2008; Mkoma, 2008; Aggarwal and Kawamura, 2009; Mkoma et al., 2009; de Souza et al., 2010; Li et al., 2010; Martin et al., 2010; Radhi et al., 2010; Weinstein et al., 2010; Batmunkh et al., 2011; Gioda et al., 2011; Pathak et al., 2011; F. Zhang et al., 2012; Cho and Park, 2013; Zhao et al., 2013; Wang et al., 2019; Kuzu et al., 2020). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.7 | Distribution of PM2.5composition mass concentration (in μg m–3) for the major PM2.5aerosol components. Those aerosol components are sulphate, nitrate, ammonium, sodium, chloride, organic carbon and elemental carbon. The central world map depicts the intermediate-level regional breakdown of observations (10 regions) following the IPCC Sixth Assessment Report Working Group III (AR6 WGIII). Monthly averaged PM2.5 aerosol component measurements are from: (i) the Environmental Protection Agency (EPA) network which include 211 monitor sites primarily in urban areas of North America during 2000–2018 (Solomon et al., 2014), (ii) the Interagency Monitoring of Protected Visual Environments (IMPROVE) network during 2000–2018 over 198 monitoring sites representative of the regional haze conditions over North America, (iii) the European Monitoring and Evaluation Programme (EMEP) network over 70 monitoring in Europe and (eastern) Eurasia during 2000–2018, (iv) the Acid Deposition Monitoring Network in Eastern Asia (EANET) network with 39 (18 remote, 10 rural, 11 urban) sites in Eurasia, Eastern Asia, South East Asia and Developing Pacific, and Asia-Pacific Developed during 2001–2017, (v) the global Surface Particulate Matter Network (SPARTAN) during 2013–2019 with sites primarily in highly populated regions around the world (i.e., North America, Latin America and Caribbean, Africa, Middle East, Southern Asia, Eastern Asia, South East Asia and Developing Pacific; Snider et al., 2015, 2016), and (vii) individual observational field campaign averages over Latin America and Caribbean, Africa, Europe, Eastern Asia, and Asia-Pacific Developed (Celis et al., 2004; Feng et al., 2006; Bourotte et al., 2007; Fuzzi et al., 2007; Mariani and de Mello, 2007; Molina et al., 2007, 2010; Favez et al., 2008; Mkoma, 2008; Aggarwal and Kawamura, 2009; Mkoma et al., 2009; de Souza et al., 2010; Li et al., 2010; Martin et al., 2010; Radhi et al., 2010; Weinstein et al., 2010; Batmunkh et al., 2011; Gioda et al., 2011; Pathak et al., 2011; F. Zhang et al., 2012; Cho and Park, 2013; Zhao et al., 2013; Wang et al., 2019; Kuzu et al., 2020). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). Remote-sensing instruments provide a larger-scale view of aerosol distributions and trends than ground-based monitoring networks by retrieving the aerosol optical depth (AOD), which is indirectly related to aerosol mass concentrations. AOD is the column-integrated measure of extinction of the solar intensity due to aerosols at a given wavelength, and is therefore relevant to the estimation of the radiative forcing of aerosol–radiation interactions (Section 7.3.3.1). Models participating in Phase III of the AeroCom intercomparison project were found to underestimate present-day AOD by about 20% (Gliß et al., 2021), although different remote-sensing estimates obtain different estimates of global mean AOD. Gliß et al. (2021) also highlight the considerable diversity in the simulated contribution of various aerosol types to total AOD. However, models simulate regional trends in AODs that agree well, when expressed as percentage change, with ground- (Mortier et al. , 2020; Gliß et al. , 2021) and satellite-based (Cherian and Quaas, 2020; Gliß et al., 2021) observations. AOD trends simulated by CMIP6 models are more consistent with satellite-derived trends than CMIP5 models for several sub-regions, thanks to improved emissions estimates (Cherian and Quaas, 2020).
All CMIP6 models simulate a positive trend in global mean AOD from 1850, with a strong increase after the 1950s coinciding with the massive increase in anthropogenic SO2 emissions (Figure 6.8). Global mean AOD increases have slowed since 1980, or even reversed in some models, as a result of a compensation between SO2 emissions decreases over the USA and Europe in response to air-quality controls since the mid-1980s, and increases over Asia. From about 2000, global mean AOD stabilized in the models, driven by soaring emissions in Southern Asia and declining emissions in Eastern Asia (Section 6.2.1). Trends after around 2010 are difficult to assess from CMIP6 models because the historical simulations end at 2014. Nevertheless, the strong decline in anthropogenic SO2 emissions over Eastern Asia since 2011 is underestimated in the CMIP6 emissions database (Hoesly et al., 2018), indicating that the observed AOD change over Eastern Asia may not be captured accurately by CMIP6 models (Wang et al., 2021). While all CMIP6 models simulate the increase of AOD between 1850 and 2014 there is strong inter-model diversity in the simulated AOD change since 1850 ranging from 0.01 (15%) to 0.08 (53%) in 2014. Some models therefore lie outside the 68% confidence interval of 0.02 (15%) to 0.04 (or 30%) for global AOD change in 2005–2015 compared to 1850, estimated by Bellouin et al. (2020) based on observational and model (excluding CMIP6) lines of evidence. In addition to the horizontal distribution of aerosols documented by AOD, their number size distribution, vertical distribution, optical properties, hygroscopicity, ability to act as CCN, chemical composition, mixing state and morphology are key elements to assess their climate effect (Section 6.4).
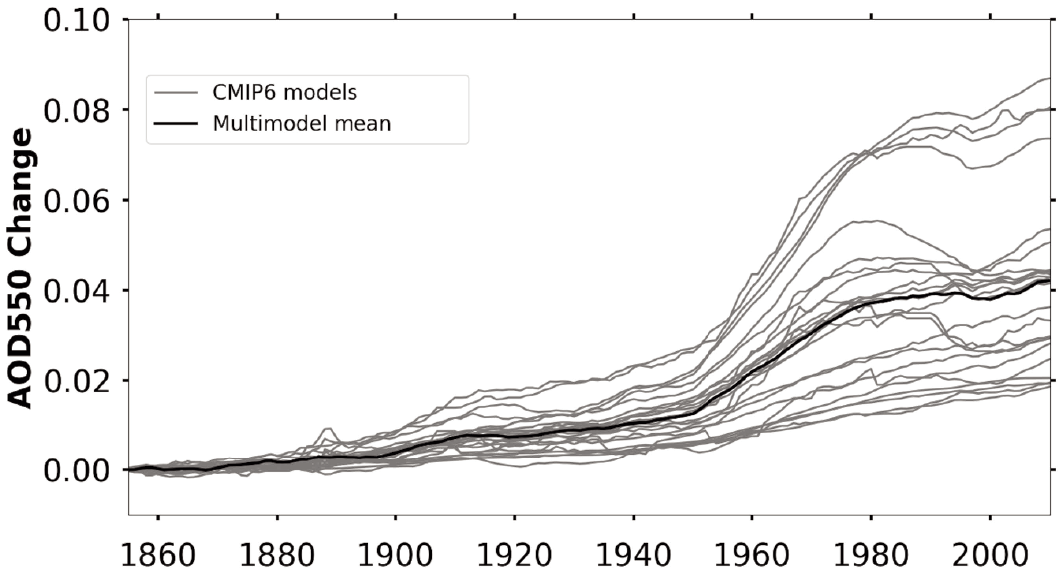 Figure 6.8 | Time evolution of changes in global mean aerosol optical depth (AOD) at 550 nm. The year of reference is 1850. Data are shown from individual Coupled Model Intercomparison Project Phase 6 (CMIP6) historical simulations. Each time series corresponds to the ensemble mean of realizations done by each model. Simulation results from years including major volcanic eruptions (e.g., Novarupta, 1912; Pinatubo, 1991), are excluded from the analysis for models encompassing the contribution of stratospheric volcanic aerosols to total AOD. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.8 | Time evolution of changes in global mean aerosol optical depth (AOD) at 550 nm. The year of reference is 1850. Data are shown from individual Coupled Model Intercomparison Project Phase 6 (CMIP6) historical simulations. Each time series corresponds to the ensemble mean of realizations done by each model. Simulation results from years including major volcanic eruptions (e.g., Novarupta, 1912; Pinatubo, 1991), are excluded from the analysis for models encompassing the contribution of stratospheric volcanic aerosols to total AOD. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). 6.3.5.1 Sulphate (SO42–)
Sulphate aerosols (or sulphate-containing aerosols) are emitted directly or formed in the atmosphere by gas- and aqueous-phase oxidation of precursor sulphur gases, including SO2, DMS and carbonyl sulphide (OCS), emitted from anthropogenic and natural sources (Section 6.2). Sulphate aerosols influence climate forcing directly by either scattering solar radiation or absorbing longwave radiation, and indirectly by influencing cloud micro- and macrophysical properties and precipitation (Boucher et al., 2013; Myhre et al., 2013). Additionally, sulphate aerosols and sulphate deposition have a large impact on air quality and ecosystems (Reis et al., 2012). The majority of sulphate particles are formed in the troposphere, however, SO2 and other longer-lived natural precursors, such as OCS, transported into the stratosphere, contribute to the background stratospheric aerosol layer (Kremser et al., 2016). SO2 emissions from volcanic eruptions are a significant source of stratospheric sulphate loading (see Chapter 2 for reconstruction of stratospheric aerosol optical depth and Chapter 7 for radiative forcing of volcanic aerosols). Furthermore, studies suggest sulphate contributions from anthropogenic SO2 emissions transported into the stratosphere could have a consequent impact on radiative forcing (Myhre et al., 2004; Yu et al., 2016). However, there is significant uncertainty in the relative importance of this stratospheric sulphate source (Kremser et al., 2016).
Process understanding of sulphate production pathways from SO2 emissions has seen some progress since AR5. More specifically, many global climate models now have a more complete description of chemical reactions such that oxidant levels (including ozone) are better described, include a pH-dependence of SO2 oxidation (e.g., Kirkevåg et al., 2018; Bauer et al., 2020), and implement explicit descriptions of ammonium and nitrate aerosol components, which may influence the partitioning of sulphate (Bian et al. , 2017; Lund et al. , 2018a). The pH influences the heterogeneous chemistry as well as the physical properties of the aerosols, and this topic has been a subject of growing interest since AR5 (Cheng et al., 2016; Freedman et al., 2019; Nenes et al., 2020). Increases in cloudwater pH have been shown to significantly increase the radiative forcing due to sulphate aerosols (Turnock et al., 2019).
Sulphate is removed from the atmosphere by dry deposition and wet scavenging, and these processes depend on the characteristics of the Earth’s surface, and the intensity, frequency and amount of precipitation (Boucher et al., 2013). Even though there have been some improvements since AR5, representation of atmospheric transport and of wet scavenging and related cloud processes remains a key source of uncertainty in the simulated aerosol distribution and lifetime, with further consequences for the sulphate forcing estimates (Kristiansen et al., 2016; Lund et al., 2018a). There are also still relatively large uncertainties in the emission height used in models affecting the simulated aerosol distribution (Yang et al. , 2019a).
Based on long-term surface-based in situ observations, AR5 reported a strong decline in sulphate aerosols in Europe and the USA over 1990–2009, with the largest decreases occurring before 2000 in Europe and post-2000 in the USA. Since AR5, atmospheric measurements in conjunction with model results have provided insights into the spatial and temporal distribution of sulphate and sulphur deposition (Vet et al., 2014; Tan et al., 2018; Aas et al., 2019). The in situ observations in North America and Europe reveal substantial reduction since the measurements started around 1980, though the trends have not been linear through this period (Table 6.5). Several regional studies agree with these trend estimates for Europe (Banzhaf et al. , 2015; Theobald et al. , 2019) and North America (Sickles II and Shadwick, 2015; Paulot et al. , 2016). Further, the concentrations of primary emitted SO2 (Section 6.3.3.5) show greater decreases than secondary sulphate aerosols over these regions due to a combination of higher oxidation rate (hence more SO2 converted to SO42–) and increased dry deposition rate of SO2 (Fowler et al. , 2009; Banzhaf et al. , 2015). In situ observations over other parts of the world are scattered (Figure 6.7), and the lack of observations makes it too uncertain to quantify regional representative trends (Hammer et al., 2018). However, limited in situ observations in Eastern Asia indicate an increase in atmospheric sulphate up to around 2005 and then a decline (Aas et al., 2019), which is confirmed by satellite observations of SO2 (Section 6.3.3.5). In India, on the other hand, satellite observations indicate a rapid increase in the SO2 levels (Krotkov et al., 2016), and long-term measurements of sulphate in precipitation in India further provide evidence of an increasing trend from 1980–2010 (Bhaskar and Rao, 2017; Aas et al., 2019). Further improvements in global trend assessments are expected with new integrated reanalysis products from the Earth-system data assimilation projects (Randles et al., 2017; Inness et al., 2019).
Indirect evidence of decadal trends in the atmospheric loading of sulphur are provided by Alpine ice cores, mainly influenced by European sources (Engardt et al., 2017), and ice cores from Svalbard (Samyn et al., 2012) and Greenland (Patris et al., 2002; Iizuka et al., 2018) influenced by sources in Europe and North America. These show similar patterns with a weak increase from the end of the 19th century up to around 1950, followed by a steep increase up to around 1980, and then a significant decrease over the next two decades (see also Section 2.2.6). This general trend is consistent with the emissions of SO2 in North America and Europe (Figures 6.18 and 6.19; Hoesly et al. , 2018).
Global and regional models qualitatively reproduce observed trends over North America and Europe for the period 1990–2015 for which emissions changes are generally well quantified (Aas et al., 2019; Mortier et al., 2020), building confidence in the relationship between emissions, concentration, deposition and radiative forcing derived from these models. However, the models seem to systematically underestimate sulphate (Bian et al., 2017; Lund et al., 2018a) and AOD (Lund et al., 2018a; Gliß et al., 2021), and there are quite large differences in the models’ distribution of the concentration fields of sulphate driven by differences in the representation of photochemical production and sinks of aerosols. One global model study also highlighted biases in simulated sulphate trends over the 2001–2015 period over eastern China due to uncertainties in the CEDS anthropogenic SO2 emissions trends (Paulot et al. , 2018a).
In summary, there is high confidence that the global tropospheric sulphate burden increased from 1850 to around 2005, but there are large regional differences in the magnitude. Sulphate aerosol concentrations in North America and Europe have declined over 1980–2015 with slightly stronger reductions in North America (47 ± 20%) than in Europe (40 ± 30%) over 2000–2015, though Europe had larger reductions in the prior decade (1990–2000; 52 ± 21% and 21 ± 14% respectively for Europe and North America). In Asia, the trends are more scattered, though there is medium confidence that there was a strong increase up to around 2005, followed by a steep decline in China, while over India, the concentrations are increasing steadily.
6.3.5.2 Ammonium (NH4+) and Nitrate Aerosols (NO3–)
Ammonium sulphate and ammonium nitrate aerosols are formed when NH3 reacts with nitric acid (HNO3) and sulphuric acid (H2SO4), produced in the atmosphere by the oxidation of NOx and SO2 respectively. Ammonium nitrate is formed only after H2SO4 is fully neutralized. NH4+ and NO3–aerosols produced via these gas-to-particle reactions are a major fraction of fine-mode particles (with diameter <1µm) affecting air quality and climate. Coarse-mode nitrate, formed by the heterogeneous reaction of nitric acid with dust and sea salt, dominates the overall global nitrate burden, but has little radiative effect (Hauglustaine et al., 2014; Bian et al., 2017). Trends in ammonium (NH4+) and nitrate (NO3–) were not assessed in AR5.
Global model present-day estimates of the global NH4+burden range from 0.1–0.6 TgN (Bian et al., 2017). Models generally simulate surface NH4+ concentrations better than surface NH3 concentrations (Bian et al., 2017), which reflects its thermodynamic control by SO42–rather than NH3 (Shi et al., 2017). The concomitant increases of NH3, SO2, and NOx emissions (see Section 6.2) have led to a factor of three to nine increase in the simulated NH4+burden from 1850–2000 (Hauglustaine et al., 2014; Lund et al., 2018a), driven primarily by ammonium sulphate (70–90%). The increases in the NH3 and NH4+burdens are indirectly supported by the observed increase of NH4+ concentration in ice cores in mid- to high latitudes (Kang et al. , 2002; Kekonen et al. , 2005; Lamarque et al. , 2013; Iizuka et al. , 2018).
Ammonium nitrate is semi-volatile, which results in complex spatial and temporal patterns in its concentrations (Putaud et al., 2010; Hand et al., 2012a; H. Zhang et al., 2012), reflecting variations in its precursors, NH3 and HNO3, as well as SO42–, non-volatile cations, temperature and relative humidity (Nenes et al. , 2020). High relative humidity and low temperature as well as elevated fine particulate matter loading (Huang et al. , 2014; Petit et al. , 2015; H. Li et al. , 2016; Sandrini et al. , 2016) favour nitrate production. Measurements reveal the high contribution of NO3– to surface PM2.5 (>30%) in regions with elevated regional NOx and NH3 emissions, such as the Paris area (Beekmann et al., 2015; Zhang et al., 2019), northern Italy (Masiol et al., 2015; Ricciardelli et al., 2017), Salt Lake City (Kuprov et al., 2014; Franchin et al., 2018), the North China Plains (Guo et al., 2014; Chen et al., 2016) and New Delhi(Pant et al., 2015). Recent observations also show that ammonium nitrate contributes to the Asian tropopause aerosol layer (Vernier et al., 2018; Höpfner et al., 2019). Model diversity in simulating the present-day global fine-mode NO3–burden is large with two multi-model intercomparison studies reporting estimates in the range of 0.14–1.88 Tg and 0.08–0.93 Tg respectively (Bian et al., 2017; Gliß et al., 2021). Models differ in their estimates of the global tropospheric nitrate burden by up to a factor of 13 with differences remaining nearly the same across the CMIP5 and CMIP6 generation of models (Bian et al., 2017; Gliß et al., 2021). While regional patterns in the concentration of fine-mode NO3– are qualitatively captured by models, the simulation of fine-mode NO3–is generally worse than that of NH4+or SO42– (Bian et al., 2017). This can be partly attributed to the semi-volatile nature of ammonium nitrate and biases in the simulation of its precursors (Heald et al., 2014; Paulot et al., 2016), including the sub-grid scale heterogeneity in NOx and NH3 emissions (Zakoura and Pandis, 2018).
Models indicate that the burden of fine-mode NO3–has increased by a factor of two to five from 1850–2000 (Xu and Penner, 2012; Hauglustaine et al., 2014; Lund et al., 2018a), an increase that has accelerated between 2001 and 2015 (Lund et al., 2018a; Paulot et al., 2018b). The sensitivity of NO3– to changes in NH3, SO42–, and HNO3 is determined primarily by aerosol pH, temperature, and aerosol liquid water (Guo et al., 2016, 2018; Weber et al., 2016; Nenes et al., 2020). In regions where aerosol pH is high, changes in NO3–follow changes in NOx emissions, consistent with the observed increase of ammonium nitrate in northern China from 2000–2015 (Wen et al., 2018) and its decrease in the US Central Valley (Pusede et al., 2016). In contrast, the decrease in SO2 emissions in the south-east USA has caused little change in NO3– from 1998–2014 as nitric acid largely remains in the gas phase due to highly acidic aerosols (Weber et al., 2016; Guo et al., 2018).
In summary, there is high confidence that the NH4+ and NO3–burdens have increased from the pre-industrial period to the present day, although the magnitude of the increase is uncertain especially for NO3–. The sensitivity of NH4+ and NO3– to changes in NH3, H2SO4 and HNO3 is well understood theoretically. However, it remains challenging to represent in models, in part because of uncertainties in the simulation of aerosol pH, and only a minority of ESMs consider nitrate aerosols in CMIP6.
6.3.5.3 Carbonaceous Aerosols
Carbonaceous aerosols are black carbon (BC)3, which is soot made almost purely of carbon, and organic aerosols4 (OA), which also contain hydrogen and oxygen and can be of both primary (POA) or secondary (SOA) origin. BC and a fraction of OA called brown carbon (BrC) absorb solar radiation. The various components of carbonaceous aerosols have different optical properties, so the knowledge of their partition, mixing, coating and ageing is essential to assess their climate effect (Section 7.3.3.1.2).
Carbonaceous aerosols receive attention in the scientific and policy arena due to their radiative forcing, and their sizeable contribution to PM in an air-quality context (Rogelj et al. , 2014b; Harmsen et al. , 2015; Shindell et al. , 2016; Haines et al. , 2017; Myhre et al. , 2017). BC exerts a positive forcing, but the forcing from carbonaceous aerosol as a whole is negative (Bond et al., 2013; Thornhill et al., 2021b). On average, carbonaceous aerosols account for 50–70% of PM with a diameter lower than 1 µm in polluted and pristine areas (Zhang et al. , 2007; Carslaw et al. , 2010; Andreae et al. , 2015; Monteiro dos Santos et al. , 2016; Chen et al. , 2017).
An extensive review on BC (Bond et al., 2013) discussed limitations in inferring its atmospheric abundance and highlighted inconsistencies between different terminology and related measurement techniques (Petzold et al., 2013; Sharma et al., 2017). Due to a lack of global observations, AR5 only reported declining total carbonaceous aerosol trends from the USA and a declining BC trend from the Arctic based on data available up to 2008. Since AR5, the number of observation sites has grown worldwide (Figure 6.7) but datasets suitable for global trend analyses remain limited (Reddington et al., 2017; Laj et al., 2020). Locally, studies based on observations from rural and background sites have reported decreasing surface carbonaceous aerosol trends in the Arctic, Europe, the USA, Japan and India (Table 6.6). Increases in carbonaceous aerosol concentrations in some rural sites of the western USA have been associated with wildfires (Hand et al., 2013; Malm et al., 2017). Long-term OA observations are scarce, so their trends outside of the USA are difficult to assess. Ice-core analysis has provided insight into carbonaceous aerosol trends predating the satellite and observation era over the Northern Hemisphere (Section 2.2.6, Figure 2.9b).
Species | Analysis Period | Change/Trends | References |
BC | 1990–2009 | Arctic Sites (Alert, Barrow, Ny-Alesund) −2% yr−1 | |
1970–2010 | Finland (Kevo remote site) −1.8% yr–1 | ||
2005–2014 | Germany (rural site) −2% yr–1 | ||
2009–2016 | United Kingdom (Harwell rural site) −8% yr–1 | ||
2009–2019 | Japan (Fukue Island) −5.8 ± 1.5% yr–1 | ||
2009–2015 | India (Darjeeling mountain site) −5% yr–1 | ||
OA | 2001–2015 | USA (IMPROVE sites east of 100°W) −2% yr–1 | |
Total Carbon (EC + OC) | 1990–2010 | USA (IMPROVE sites) Western USA: −4 to −5% yr–1 Eastern USA: −1 to −2% yr–1 | |
2002–2010 | Spain (Montseny rural site) −5% yr–1 |
Knowledge of carbonaceous aerosol atmospheric abundance continues to rely on global models due to a lack of global-scale observations. For BC, models agree within a factor of two with measured surface mass concentrations in Europe and North America, but underestimate concentrations at the Arctic surface by one to two orders of magnitude, especially in winter and spring (Lee et al., 2013; Lund et al., 2018a). For OA, AeroCom models underestimate surface mass concentrations by a factor of two over urban areas, as their low horizontal resolution prevents them from resolving local pollution peaks (Tsigaridis et al., 2014; Lund et al., 2018a). Models agree within a factor of two with OA surface concentrations measured at remote sites, where surface concentrations are more spatially uniform (Tsigaridis et al., 2014).
BC and OA lifetimes are estimated to be 5.5 days ± 35% and 6.0 days ± 29% (median ± 1 standard deviation), respectively, based on an ensemble of 14 models (Gliß et al., 2021). Disagreement in simulated lifetime leads to horizontal and vertical variations in predicted carbonaceous aerosol concentrations, with implications for radiative forcing (Samset et al., 2013; Lund et al., 2018b). Airborne campaigns have provided valuable vertical-profile measurements of carbonaceous aerosol concentrations (Schwarz et al. , 2013; Freney et al. , 2018; Hodgson et al. , 2018; Schulz et al. , 2019; D. Zhao et al. , 2019; Morgan et al. , 2020). Compared to those measurements, models tend to transport BC too high in the atmosphere, suggesting that lifetimes are not larger than 5.5 days (Samset et al., 2013; Lund et al., 2018b). Newly developed size-dependent wet-scavenging parametrization for BC (Taylor et al., 2014; Schroder et al., 2015; Ohata et al., 2016; G. Zhang et al., 2017; Ding et al., 2019; Moteki et al., 2019; Motos et al., 2019) may lead to decreased BC lifetimes and improve agreement with observed vertical profiles.
Simulated BC burdens show a large spread among models (Gliß et al., 2021), despite using harmonised primary emissions, because of differences in BC removal efficiency linked to different treatment of ageing and mixing, particularly in strong source regions. The multi-model median BC burden for the year 2010 from Gliß et al. (2021), based on 14 AeroCom models, is 0.131 ± 0.047 Tg (median ± 1 standard deviation). The range encompasses values reported by independent single-model estimates (Huang et al. , 2013; Lee et al. , 2013; Sharma et al. , 2013; Q. Wang et al. , 2014; Tilmes et al. , 2019).
Simulated OA burdens also show a large spread among global models, with Gliß et al. (2021) reporting a multi-model median of 1.91 ± 0.65 Tg for the year 2010. The large spread reflects the wide range in the complexity of the OA parametrizations, particularly for SOA formation, as well as in the primary OA emissions (Tsigaridis et al., 2014; Gliß et al., 2021). The uncertainties are particularly large in model estimates of SOA production rates, which vary between 10 and 143 Tg yr–1 (Tsigaridis et al., 2014; Hodzic et al., 2016; Tilmes et al., 2019). While the level of complexity in the representation of OA in global models has increased since AR5 (Shrivastava et al., 2017; Hodzic et al., 2020), limitations in process-level understanding of the formation, ageing and removal of organic compounds lead to uncertainties in the global model predictions of global OA burden and distribution as well as the relative contribution of POA and SOA to OA. Jo et al. (2016) estimated that BrC contributes about 20% of total OA burden. That would give BrC a burden similar to that of BC (low confidence), enhancing the overall forcing exerted by carbonaceous aerosol absorption (Zhang et al., 2020).
In summary, the lack of global-scale observations of carbonaceous aerosols, the complexity of processes influencing them, and the large spread in their simulated global budget and burdens means that there is only low confidence in the quantification of the present-day atmospheric distribution of individual components of carbonaceous aerosols. Global trends in carbonaceous aerosols cannot be characterized due to limited observations, but sites representative of background conditions have reported multi-year declines in BC over several regions of the Northern Hemisphere.
6.3.6 Implications of SLCF Abundances for Atmospheric Oxidizing Capacity
The atmospheric oxidising capacity is determined primarily by tropospheric hydroxyl (OH) radical and to a smaller extent by NO3 radical, ozone, hydrogen peroxide (H2O2) and halogen radicals. OH is the main sink for many SLCFs, including methane, halogenated compounds (HCFCs and HFCs), CO and NMVOCs, controlling their lifetimes and consequently their abundance and climate influence. OH-initiated oxidation of methane, CO and NMVOCs in the presence of NOx leads to the production of tropospheric ozone. OH also contributes to the formation of aerosols from oxidation of SO2 to sulphate and NMVOCs to secondary organic aerosols. The evolution of the atmospheric oxidising capacity of the Earth driven by human activities and natural processes is, therefore, of significance for climate and air-quality concerns.
The main source of tropospheric OH is the photoexcitation of tropospheric ozone that creates an electronically excited oxygen atom which reacts with water vapour producing OH. A secondary source of importance for global OH is the recycling of peroxy radicals formed by the reaction of OH with reduced and partly oxidized species, including methane, CO and NMVOCs. In polluted air, NOx emissions control the secondary OH production, while in pristine air it occurs via other mechanisms involving, in particular, isoprene (Lelieveld et al., 2016; Wennberg et al., 2018). Knowledge of the effect of isoprene oxidation on OH recycling has evolved tremendously over the past decade, facilitating mechanistic explanation of elevated OH concentrations observed in locations characterised by low NOx levels (Hofzumahaus et al. , 2009; Paulot et al. , 2009; Peeters et al. , 2009, 2014; Fuchs et al. , 2013). Since AR5, the inclusion of improved chemical mechanisms in some CTMs suggest advances in understanding of the global OH budget, however, these improvements have yet to be incorporated in CMIP6-generation ESMs.
As a result of the complex photochemistry, tropospheric OH abundance is sensitive to changes in SLCF emissions as well as climate. Increases in methane, CO and NMVOCs reduce OH while increases in water vapour and temperature, incoming solar radiation, NOx and tropospheric ozone enhance OH. The OH level thus responds to climate change and climate variability via its sensitivity to temperature and water vapour, as well as the influence of climate on natural emissions (e.g., wetland methane emissions, lightning NOx, BVOCs, fire emissions) with consequent feedbacks on climate (Section 6.4.5). Climate modes of variability, like El Niño–Southern Oscillation, also contribute to OH variability via changes in lightning NOx emissions and deep convection (Turner et al. , 2018), and fire emissions (Rowlinson et al., 2019).
Global-scale OH observations are non-existent because of its extremely short lifetime (around 1 second) and therefore global OH abundance and its time variations are either inferred from atmospheric measurements of methyl chloroform (MCF; Prinn et al., 2018 and references therein) or derived from global atmospheric chemistry models (Lelieveld et al., 2016). The AR5 reported small interannual OH variations in the 2000s based on atmospheric inversions of MCF observations (within ±5%) and global CCMs and CTMs (within ±3%) (Ciais et al., 2013).
Since AR5, there is much closer agreement in the estimates of interannual variations in global mean OH derived from atmospheric inversions, empirical reconstruction, and global CCMs and ESMs, with an estimate of 2–3% over the 1980–2015 period (Table 6.7). While the different methodologies agree on the occurrence of small interannual variations, there is much debate over the longer-term global OH trend. Two studies using multi-box model inversions of MCF and methane observations suggest large positive and negative trends since the 1990s in global mean OH (Rigby et al., 2017;Turner et al., 2017), however, both find that observational constraints are weak, such that a wide range of multi-annual OH variations are possible. Indeed, Naus et al. (2019) find an overall positive global OH trend over the past two decades (Table 6.7) after accounting for uncertainties and biases in atmospheric MCF and methane inversions, confirming the weakness in observational constraints for deriving OH trends. Global ESMs, CCMs and CTMs exhibit increasing global OH after 1980 contrary to the lack of trend derived from some atmospheric inversions and empirical reconstructions (Table 6.7). In particular, a three-member ensemble of ESMs participating in the AerChemMIP/CMIP6 agrees that global OH has increased since 1980 by around 9% (Figure 6.9) with an associated reduction in methane lifetime (Stevenson et al., 2020). This positive OH trend is in agreement with the OH increase of about 7% derived by assimilating global-scale satellite observations of CO over the 2002–2013 period (with CO declining trends) into a CCM (Section 6.3.4; Gaubert et al., 2017). Multi-model sensitivity analysis suggests that increasing OH since 1980 is predominantly driven by changes in anthropogenic SLCF emissions with the complementary influence of increasing NOx and decreasing CO emissions (Stevenson et al., 2020).
Reference | Time Period | OH Trends and IAV | Approach |
Inversion and Empirical Methods Based on Observations | |||
1998–2007 | 2.3 ± 1.3% (IAV) | 3D inversion | |
2000s | within ±5% (IAV) | AR5 based on inversions | |
1993–2011 | ±2.3% (IAV) | Box-model inversion | |
1980–2014 | 10% increase from the late 1990s–2004; 10% decrease from 2004–2014 | Box-model inversion | |
1983–2015 | about 7% increase in 1991–2001; 7% decrease in 2003–2016 | Box-model inversion | |
1980–2015 | 1.6% (IAV) | Empirical reconstruction | |
2003–2015 | 1.8 ± 0.4% decrease | 3D inversion | |
1994–2015 | 3.8 ± 3.2% increase | Box-model inversion | |
1996–2015 | 2–3% IAV, no trend | 3D inversion | |
Global CTMs, CCMs and ESMs | |||
1860–2005 1980–2000 | 6% decrease About 3% increase | CCM | |
1997–2009 | 0.7–1.1% (IAV) | Multi-model CTMs | |
2000s | within ±3% (IAV) | AR5 based on CCMs | |
1998–2006 | increasing trend | 3D CTM | |
1980–2000 1850–2000 | 3.5 ± 2.2 % increase −0.6 ± 8.8% | Multi-model CCMs/CTMs | |
1770s–1990s | 5.3% increase | CCM | |
1970–2012 | 8% increase | 3D CTM | |
2002–2013 | 7% increase | CCM with assimilated satellite CO observations | |
1960–2010 1980–2000 | 1.9 ± 1.2 % (IAV) 4.6 ± 2.4 % increase | Multi-model CCMs/CTMs | |
1980–2014 1850–1980 | 9% increase no trend | Multi-model ESMs | |
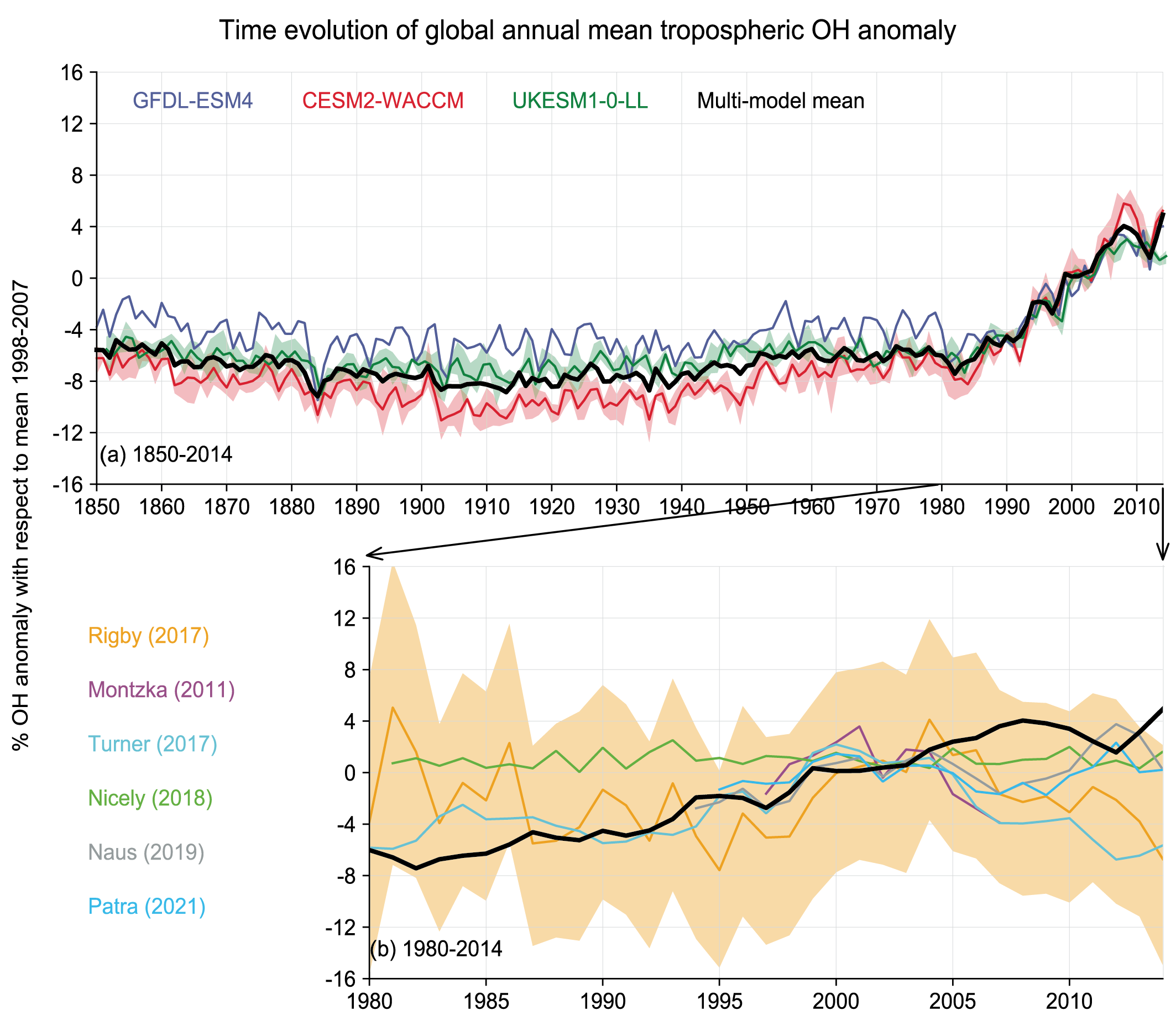 Figure 6.9 | Time evolution of global annual mean tropospheric hydroxyl (OH) over the historical period, expressed as a percentage anomaly relative to the mean over 1998–2007. (a) Results from three CMIP6 models, including UKESM1-0LL (green), GFDL-ESM4 (blue), and CESM2-WACCM (red), are shown; the shaded light green and light red bands show mean over multiple ensemble members for UKESM1-0LL (3) and CESM2-WACCM (3) models, respectively with the multi-model mean anomalies shown in thick black line. (b) Multi-model mean OH anomalies for the 1980–2014 period compared with those derived from observational-based inversions from Montzka et al., (2011); Rigby et al., (2017); Turner et al., (2017); Nicely et al., (2018); Naus et al., (2019); Patra et al., (2021) in the zoomed box. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.9 | Time evolution of global annual mean tropospheric hydroxyl (OH) over the historical period, expressed as a percentage anomaly relative to the mean over 1998–2007. (a) Results from three CMIP6 models, including UKESM1-0LL (green), GFDL-ESM4 (blue), and CESM2-WACCM (red), are shown; the shaded light green and light red bands show mean over multiple ensemble members for UKESM1-0LL (3) and CESM2-WACCM (3) models, respectively with the multi-model mean anomalies shown in thick black line. (b) Multi-model mean OH anomalies for the 1980–2014 period compared with those derived from observational-based inversions from Montzka et al., (2011); Rigby et al., (2017); Turner et al., (2017); Nicely et al., (2018); Naus et al., (2019); Patra et al., (2021) in the zoomed box. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). Over paleo time scales, proxy-based observational constraints from methane and formaldehyde suggest tropospheric OH to be a factor of two to four lower in the Last Glacial Maximum (LGM) relative to pre-industrial levels, though these estimates are highly uncertain (Alexander and Mickley, 2015). Global models, in contrast, exhibit no change in tropospheric OH (and consequently in methane lifetime) at the LGM relative to the pre-industrial period (Murray et al. , 2014; Quiquet et al. , 2015), however, the sign and magnitude of OH changes are sensitive to model predictions of changes in natural emissions, including lightning NOx and BVOCs, and model representation of isoprene oxidation chemistry (Achakulwisut et al., 2015; Hopcroft et al., 2017).
Regarding change since the pre-industrial era, at the time of the AR5, the ensemble mean of 17 global models participating in ACCMIP indicated little change in tropospheric OH from 1850–2000. This was due to the competing and finally offsetting changes in factors enhancing or reducing OH with a consequent small decline in methane lifetime (Naik et al., 2013; Voulgarakis et al., 2013). However, there was large diversity in both the sign and magnitude of past OH changes across the individual models attributed to the disparate implementation of chemical and physical processes (Nicely et al., 2017; Wild et al., 2020). Analysis of historical simulations from three CMIP6 ESMs indicates little change in global mean OH from 1850 to about 1980 (Stevenson et al., 2020). However, there is no observational evidence of changes in global OH since 1850 up to the early 1980s to evaluate the ESMs.
In summary, global mean tropospheric OH does not show a significant trend from 1850 up to around 1980 (low confidence). There is conflicting information from global models constrained by emissions versus observationally constrained inversion methods over the 1980–2014 period. A positive trend since 1980 (about 9% increase over 1980–2014) is a robust feature among ESMs and CCMs and there is medium confidence that this trend is mainly driven by increases in global anthropogenic NOx emissions and decreases in CO emissions. There is limited evidence and medium agreement for positive trends or absence of trends inferred from observation-constrained methods. Overall, there is medium confidence that global mean OH has remained stable or exhibited a positive trend since the 1980s.
6.4 SLCF Radiative Forcing and Climate Effects
The radiative forcing on the climate system introduced by SLCFs is distinguished from that of long-lived greenhouse gases (LLGHGs) by the diversity of forcing mechanisms for SLCFs, and the challenges of constraining these mechanisms via observations and of inferring their global forcings from available data. Chapter 7 assesses the global estimates of effective radiative forcing (ERF) due to SLCF abundance changes. This section assesses the characteristics (e.g., spatial patterns, temporal evolution) of forcings, emissions-based SLCF forcings, climate response and feedbacks due to SLCFs relying primarily on results from CMIP6 models. Additionally, the ERFs for several aerosol-based forms of solar –radiation modification (SRM) are discussed in Section 6.4.6.
Forcing and climate response due to changes in SLCFs are typically estimated from global models that vary in their representation of the various chemical, physical and radiative processes (Box 6.1) affecting the causal chain from SLCF emissions to climate response (Figure 6.1). The AR5 noted that the representation of aerosol processes varied greatly in CMIP5 models and that it remained unclear what level of sophistication is required to properly quantify aerosol effects on climate (Boucher et al., 2013). Since the AR5, Ekman (2014) found that the CMIP5 models with the most complex representations of aerosol impacts on cloud microphysics had the largest reduction in biases in surface temperature trends. CMIP6-generation CCMs that simulate aerosol and cloud-size distributions better represent the effect of a volcanic eruption on lower atmosphere clouds than a model with aerosol-mass only (Malavelle et al., 2017). This highlights the need for skilful simulation of conditions underlying aerosol–cloud interactions, such as the distribution, transport and properties of aerosol species, in addition to the interactions themselves (Chapter 7). In advance of CMIP6, representations of aerosol processes and aerosol–cloud interactions in ESMs have generally become more comprehensive (Meehl et al. , 2020; Gliß et al. , 2021; Thornhill et al. , 2021b; see also Section 1.5), with enhanced links to aerosol emissions and gas-phase chemistry. Many CMIP6 models (Annex II: Table AII.5) now simulate aerosol number size distribution, in addition to mass distribution, which is a prerequisite for accurately simulating number concentrations of CCN (Bellouin et al., 2013), while some CMIP6 models use prescribed aerosol optical properties to constrain aerosol forcing (e.g., Stevens et al., 2017). Hence, the range of complexity in aerosol modeling noted in CMIP5 is still present in the CMIP6 ensemble. Although simulated CCN have been compared to surface (Fanourgakis et al., 2019) and aircraft (Reddington et al., 2017) measurements, with mixed results, the lack of global coverage limits confidence in the evaluations. Evaluations of AOD have been more wide-ranging (Section 6.3.5; Gliß et al., 2021) but are less relevant to aerosol–cloud interactions as they do not allow the evaluation of vertical profiles, aerosol-cloud overlap regions, aerosol type or number. Nevertheless, biases in simulated patterns and trends in AOD, alongside biases in cloud fractions (Vignesh et al., 2020), likely affect quantifications of the aerosol–cloud interactions.
In summary, CMIP6 models generally represent more processes that drive aerosol–cloud interactions than the previous generation of climate models, but there is only medium confidence that those enhancements improve their fitness for the purpose of simulating radiative forcing due to aerosol–cloud interactions because only a few studies have identified the level of sophistication required to do so. In addition, the challenge of representing the small-scale processes involved in aerosol–cloud interactions, and a lack of relevant model-data comparisons, does not allow a quantitative assessment of the progress of the models from CMIP5 to CMIP6 in simulating the underlying conditions relevant for aerosol–cloud interactions at this time.
6.4.1 Historical Estimates of Regional Short-lived Climate Forcing
The highly heterogeneous distribution of SLCF abundances (Section 6.3) translates to strong heterogeneity in the spatial pattern and temporal evolution of forcing and climate responses due to SLCFs. This section assesses the spatial patterns of the current forcing due to aerosols and their historical evolution by region.
In AR5, the confidence in the spatial patterns of aerosol and ozone forcing was lower than that for the global mean because of the large spread in the regional distribution simulated by global models, and was assessed as medium. The AR5 assessment was based on aerosol and ozone RFs, and aerosol ERFs (with fixed SSTs) from ACCMIP and a small sample of CMIP5 models (Myhre et al., 2013; Shindell et al., 2013). For this assessement, the spatial distribution of aerosol ERF due to human-induced changes in aerosol concentrations over 1850–2014 is quantified based on results from a seven-member ensemble of CMIP6 ESMs including interactive gas and aerosol chemistry analysed in AerChemMIP. There is insufficient information to estimate the spatial patterns of ozone ERF from CMIP6, however, the spatial patterns in SLCF ERF are dominated by that from aerosol ERF over most regions (e.g., Shindell et al., 2015). The aerosol ERF includes contributions from both direct aerosol–radiation (ERFari) and indirect aerosol–cloud interactions (ERFaci; Section 7.3.3), and is computed as the difference between radiative fluxes from simulations with time-evolving aerosol and their precuror emissions, and identical simulations but with these emissions held at their 1850 levels (Collins et al., 2017). Both simulations are driven by time-evolving sea surface temperatures (SSTs) and sea ice from the respective coupled model historical simulation, and therefore, differ from ERFs computed using fixed pre-industrial SST and sea ice fields (Section 7.3.1), but the effect of this difference is generally small (Forster et al., 2016). A correction for land surface temperature change (Section 7.3.1) is not available from these data to explicitly quantify the contribution from adjustments. The ESMs included here used the CMIP6 anthropogenic and biomass-burning emissions for ozone and aerosol precursors but varied in their representation of the natural emissions, chemistry and climate characteristics contributing to spread in the simulated concentrations (Section 6.3) and resulting forcings, partly reflecting uncertainties in the successive processes (Thornhill et al., 2021b).
The geographical distribution of the ensemble-mean aerosol ERF over the 1850–2014 period is highly heterogeneous (Figure 6.10a) in agreement with AR5. Negative ERF is greatest over and downwind of most industrialized regions in the Northern Hemisphere and to some extent over tropical biomass-burning regions, with robust signals. The largest negative forcing occurs over Eastern Asia and Southern Asia, followed by Europe and North America, reflecting the changes in anthropogenic aerosol emissions in recent decades (Section 6.2). Positive ERF over high albedo areas, including cryosphere, deserts and clouds, also found in AR5 and attributed to absorbing aerosols, are not robust across the small CMIP6 ensemble applied here. Regionally aggregated shortwave (SW) and longwave (LW) components of the aerosol ERF exhibit similar large variability across regions (Figure 6.10b). The SW flux changes come from aerosol–radiation and aerosol–cloud interactions while the small positive LW flux changes come from aerosol–cloud interactions (related to liquid-water path changes (Section 7.3.2.2). These spatial patterns in aerosol ERF are similar to the patterns reported in AR5.
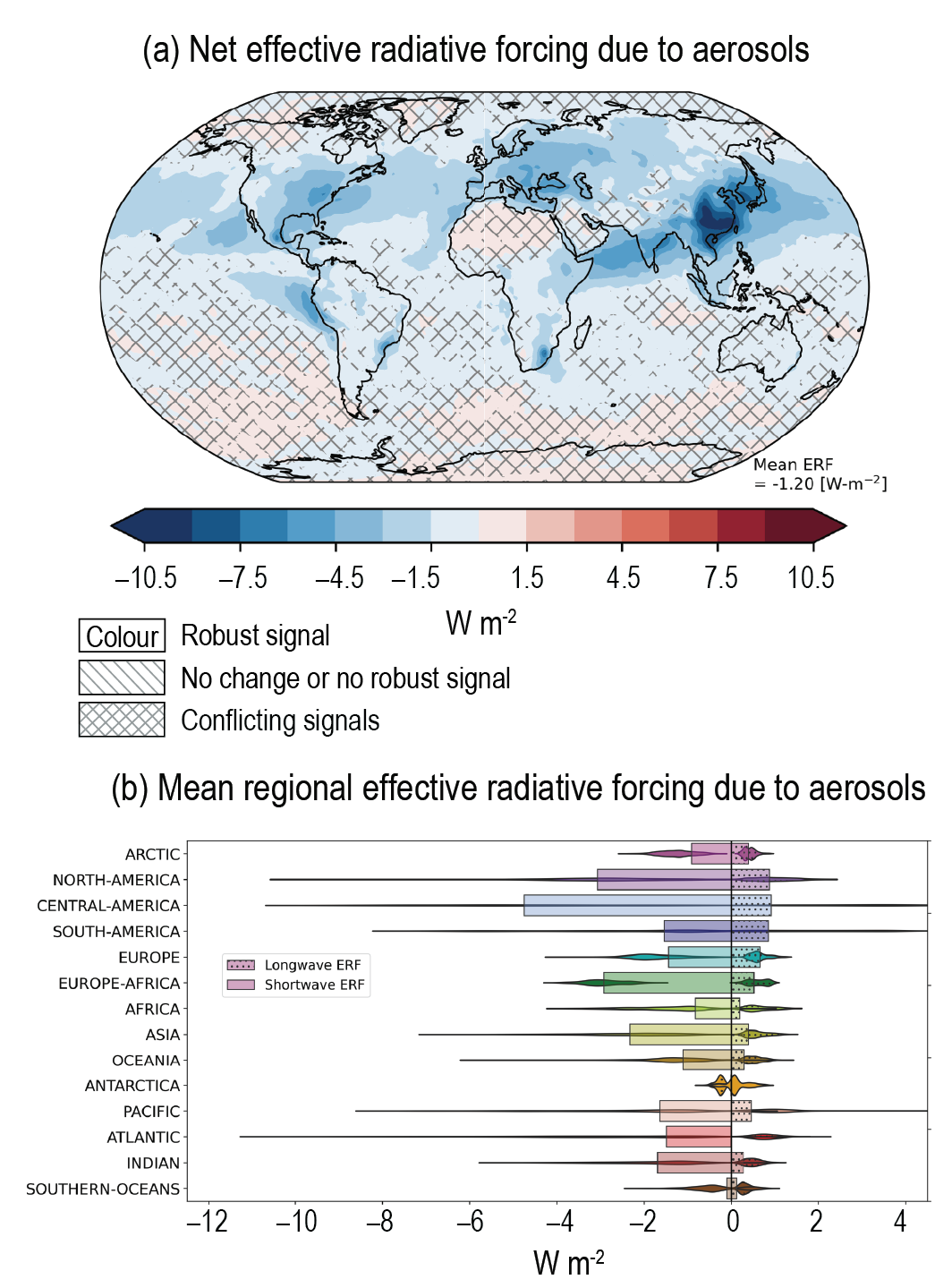 Figure 6.10 | Multi-model mean effective radiative forcings (ERFs) over the recent-past (1995-2014) induced by aerosol changes since 1850. Panel (a) shows the spatial distribution of the net ERF with area-weighted global mean ERF shown at the lower right corner. Uncertainty is represented using the advanced approach: no overlay indicates regions with robust signal, where ≥66% of models show change greater than variability threshold and ≥80% of all models agree on sign of change; diagonal lines indicate regions with no change or no robust signal, where <66% of models show a change greater than the variability threshold; crossed lines indicate regions with conflicting signal, where ≥66% of models show change greater than variability threshold and <80% of all models agree on sign of change. For more information on the advanced approach, please refer to the Cross-Chapter Box Atlas.1. Panel (b) shows the mean shortwave and longwave ERF for each of the 14 regions defined in the Atlas. Violins in panel (b) show the distribution of values over regions where ERFs are significant. ERFs are derived from the difference between top of the atmosphere (TOA) radiative fluxes for Aerosol Chemistry Model Intercomparison Project (AerChemMIP) experiments histSST and histSST-piAer (Collins et al., 2017) averaged over 1995–2014 (Box 1.4, Chapter 1). The results come from seven Earth system models: MIROC6, MPI-I-ESM-1-2-HAM, MRI-ESM2-0, GFDL-ESM4, GISS-E2-1-G, NorESM2-LM and UKESM-0-LL. These data can be seen in the Interactive Atlas. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.10 | Multi-model mean effective radiative forcings (ERFs) over the recent-past (1995-2014) induced by aerosol changes since 1850. Panel (a) shows the spatial distribution of the net ERF with area-weighted global mean ERF shown at the lower right corner. Uncertainty is represented using the advanced approach: no overlay indicates regions with robust signal, where ≥66% of models show change greater than variability threshold and ≥80% of all models agree on sign of change; diagonal lines indicate regions with no change or no robust signal, where <66% of models show a change greater than the variability threshold; crossed lines indicate regions with conflicting signal, where ≥66% of models show change greater than variability threshold and <80% of all models agree on sign of change. For more information on the advanced approach, please refer to the Cross-Chapter Box Atlas.1. Panel (b) shows the mean shortwave and longwave ERF for each of the 14 regions defined in the Atlas. Violins in panel (b) show the distribution of values over regions where ERFs are significant. ERFs are derived from the difference between top of the atmosphere (TOA) radiative fluxes for Aerosol Chemistry Model Intercomparison Project (AerChemMIP) experiments histSST and histSST-piAer (Collins et al., 2017) averaged over 1995–2014 (Box 1.4, Chapter 1). The results come from seven Earth system models: MIROC6, MPI-I-ESM-1-2-HAM, MRI-ESM2-0, GFDL-ESM4, GISS-E2-1-G, NorESM2-LM and UKESM-0-LL. These data can be seen in the Interactive Atlas. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). Time evolution of 20-year means of regional net aerosol ERF shows that the regions are divided into two groups depending on whether the mean ERF attains its negative peak value in the 1970s–1980s (e.g., Europe, North America) or in the late 1990s–2000s (e.g., Asia, South America; Figure 6.11). Qualitatively, this shift in the distribution of ERF trends is consistent with the regional long-term trends in aerosol precursor emissions (Section 6.2; Figures 6.18 and 6.19) and their abundances (Section 6.3). However, at finer regional scales, there are regions where sulphate aerosols are still following an upward trend (e.g., Southern Asia; Section 6.3.5) implying that the trends in ERF may not have shifted for these regions. The continental-scale ERF trends are also in line with the satellite-observed AOD trends assessed in Section 2.2.6. Global mean ERF reaches maximum negative values in the mid-1970s and its magnitude gradually decreases thereafter. This weakening of the negative forcing since 1990 agrees with findings that attribute this to a reduction in global mean SO2 emissions combined with an increase in global BC (Myhre et al., 2017). Uncertainties in model-simulated aerosol ERF distribution and trends can result from inter-model variations in the representation of aerosol–cloud interactions and aerosol microphysical processes as also demonstrated by Bauer et al. (2020).
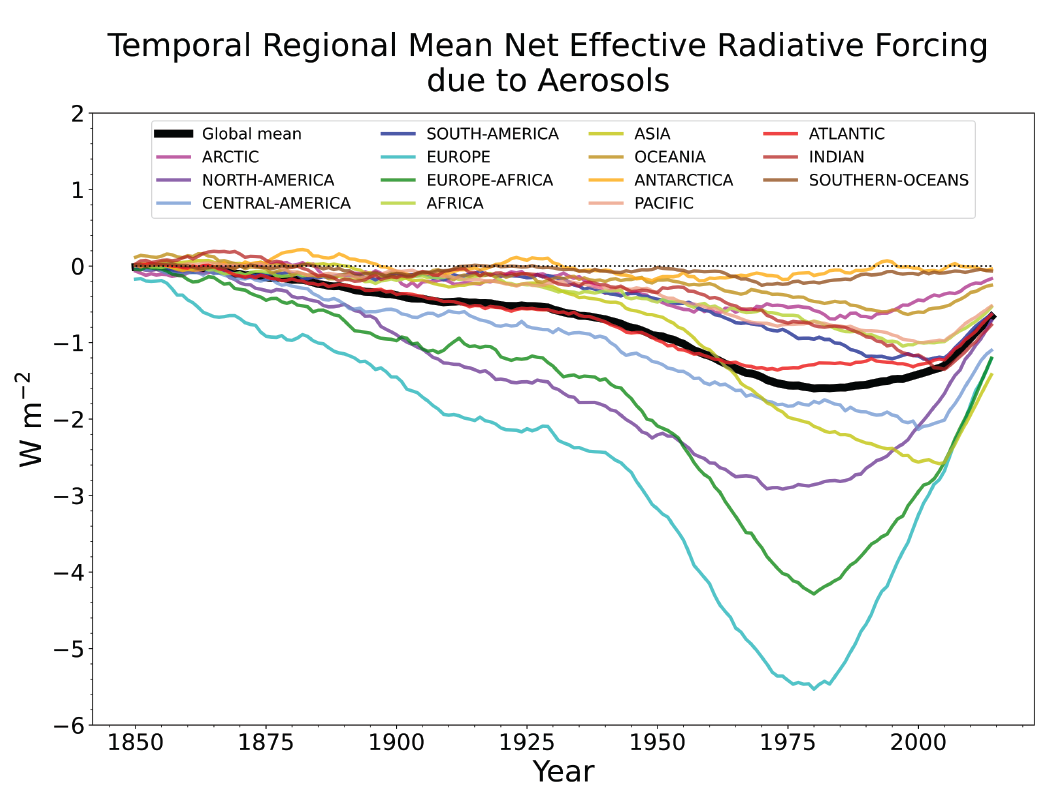 Figure 6.11 | Time evolution of 20-year multi-model mean averages of the annual area-weighted mean regional net effective radiative forcings (ERFs) due to aerosolsfor each of the 14 major regions in the Atlas, and global mean, using the models and model experiments as in Figure 6. 10. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.11 | Time evolution of 20-year multi-model mean averages of the annual area-weighted mean regional net effective radiative forcings (ERFs) due to aerosolsfor each of the 14 major regions in the Atlas, and global mean, using the models and model experiments as in Figure 6. 10. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). In summary, the spatial and temporal distribution of the net aerosol ERF from 1850–2014 is highly heterogeneous (high confidence). Globally, there has been a shift from increase to decrease of the negative net aerosol ERF driven by trends in aerosol and their precursor emissions (high confidence). However, the timing of this shift varies by continental-scale region and has not occurred for some finer regional scales.
6.4.2 Emissions-based Radiative Forcing and Effect on Global Surface Air Temperature (GSAT)
The ERFs attributable to emissions versus concentrations for several SLCFs including ozone and methane are different. A concentration change, used to assess the abundance-based ERF, results fromThe changes in emissions of multiple species and subsequent chemical reactions. The corollary is that the perturbation of a single emitted compound can induce subsequent chemical reactions and affect the concentrations of several climate forcers (chemical adjustments); this is what is accounted for in emissions-based ERF. Due to non-linear chemistry (Section 6.3) and non-linear aerosol–cloud interactions (Section 7.3.3.2), the ERF attributed to the individual species cannot be precisely defined and can only be estimated through model simulations. For example, the ERF attributed to methane emissions, which includes indirect effects through ozone formation and oxidation capacity with feedbacks on the methane lifetime, depend non-linearly on the concentrations of NOx, CO and NMVOCs. This means that the results from the model simulations depend to some extent on the chosen methodology. In AR5 (based on Shindell et al., 2009; Stevenson et al., 2013) the attribution was done by removing the anthropogenic emissions of individual species one by one from a control simulation for present-day conditions. Further, only the radiative forcings, and not the ERF (mainly including the effect of aerosol–cloud interactions) were attributed to the emitted compounds.
Since AR5, the emissions estimates have been revised and extended for CMIP6 (Hoesly et al., 2018), the models have been further developed, the period has been extended (1750–2019, versus 1750–2011 in AR5) and the experimental setup for the model simulations has changed (Collins et al., 2017), making a direct comparison of results difficult. Figure 6.12a shows the global and annual mean ERF attributed to emitted compounds over the period 1750–2019 based on AerChemMIP simulations (Thornhill et al., 2021b) where anthropogenic emissions or concentrations of individual species were perturbed from 1850 to 2014 levels (methodology described in Supplementary Material 6.SM.1).
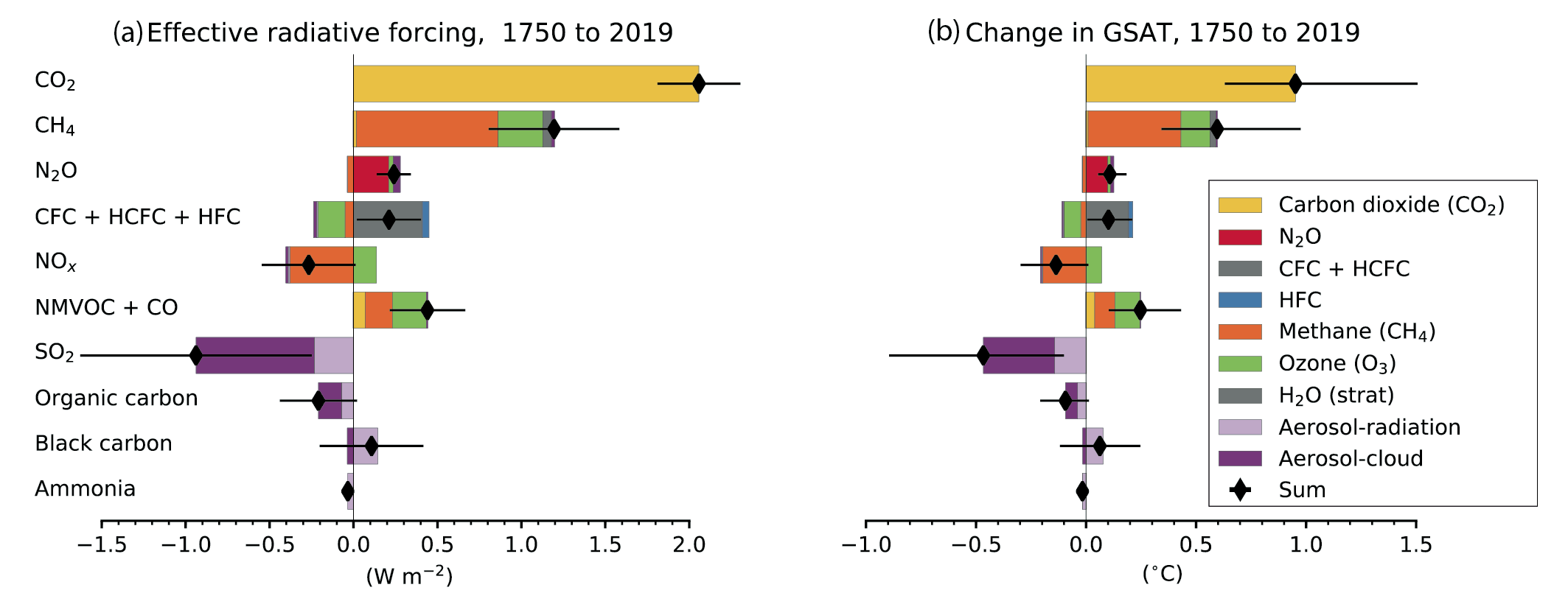 Figure 6.12 | Contribution to effective radiative forcing (ERF) (a) and global mean surface air temperature (GSAT) change (b) from component emissions between 1750 to 2019 based on CMIP6 models (Thornhill et al. , 2021b). ERFs for the direct effect of well-mixed greenhouse gases (WMGHGs) are from the analytical formulae in section 7.3.2, H2O (strat) is from Table 7.8. ERFs for other components are multi-model means from Thornhill et al. (2021b) and are based on ESM simulations in which emissions of one species at a time are increased from 1850 to 2014 levels. The derived emissions-based ERFs are rescaled to match the concentration-based ERFs in Figure 7.6. Error bars are 5–95% and for the ERF account for uncertainty in radiative efficiencies and multi-model error in the means. ERFs due to aerosol–radiation (ERFari) and cloud effects are calculated from separate radiation calls for clear-sky and aerosol-free conditions (Ghan, 2013; Thornhill et al., 2021b). ‘Cloud’ includes cloud adjustments (semi-direct effect) and ERF from indirect aerosol-cloud to –0.22 W m–2 for ERFari and –0.84 W m–2 interactions (ERFaci). The aerosol components (SO2, organic carbon and black carbon) are scaled to sum to –0.22 W m–2 for ERFari and –0.84 W m–2 for ‘cloud’ (Section 7.3.3). For GSAT estimates, time series (1750–2019) for the ERFs have been estimated by scaling with concentrations for WMGHGs and with historical emissions for SLCFs. The time variation of ERFaci for aerosols is from Chapter 7. The global mean temperature response is calculated from the ERF time series using an impulse response function (Cross-Chapter Box 7.1) with a climate feedback parameter of –1.31 W m–2°C–1. Contributions to ERF and GSAT change from contrails and light-absorbing particles on snow and ice are not represented, but their estimates can be seen on Figure 7.6 and 7.7, respectively. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.12 | Contribution to effective radiative forcing (ERF) (a) and global mean surface air temperature (GSAT) change (b) from component emissions between 1750 to 2019 based on CMIP6 models (Thornhill et al. , 2021b). ERFs for the direct effect of well-mixed greenhouse gases (WMGHGs) are from the analytical formulae in section 7.3.2, H2O (strat) is from Table 7.8. ERFs for other components are multi-model means from Thornhill et al. (2021b) and are based on ESM simulations in which emissions of one species at a time are increased from 1850 to 2014 levels. The derived emissions-based ERFs are rescaled to match the concentration-based ERFs in Figure 7.6. Error bars are 5–95% and for the ERF account for uncertainty in radiative efficiencies and multi-model error in the means. ERFs due to aerosol–radiation (ERFari) and cloud effects are calculated from separate radiation calls for clear-sky and aerosol-free conditions (Ghan, 2013; Thornhill et al., 2021b). ‘Cloud’ includes cloud adjustments (semi-direct effect) and ERF from indirect aerosol-cloud to –0.22 W m–2 for ERFari and –0.84 W m–2 interactions (ERFaci). The aerosol components (SO2, organic carbon and black carbon) are scaled to sum to –0.22 W m–2 for ERFari and –0.84 W m–2 for ‘cloud’ (Section 7.3.3). For GSAT estimates, time series (1750–2019) for the ERFs have been estimated by scaling with concentrations for WMGHGs and with historical emissions for SLCFs. The time variation of ERFaci for aerosols is from Chapter 7. The global mean temperature response is calculated from the ERF time series using an impulse response function (Cross-Chapter Box 7.1) with a climate feedback parameter of –1.31 W m–2°C–1. Contributions to ERF and GSAT change from contrails and light-absorbing particles on snow and ice are not represented, but their estimates can be seen on Figure 7.6 and 7.7, respectively. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). The ERF based on primary CO2 emissions is slightly lower than the abundance-based estimate (Section 7.3.2.1) because the abundance-based ERF combines the effect of primary CO2 emissions and a small additional secondary contribution from atmospheric oxidation of methane, CO, and NMVOCs (4%) of fossil origin, consistent with AR5 findings.
Ozone-depleting substances, such as N2O and halocarbons, cause a reduction in stratospheric ozone, which affects ozone and OH production in the troposphere through ultraviolet radiation changes (and thus affect methane). They also have indirect effects on aerosols and clouds (Karset et al., 2018), since changes in oxidants induce changes in the oxidation of aerosol precursors.
The net ERF from N2O emissions is estimated to be 0.24 [0.13 to 0.34] W m–2, which is very close to the abundance-based estimate of 0.21 W m–2Section 7.3.2.3). The indirect contributions from N2O are relatively minor with negative (methane-lifetime) and positive (ozone-and-cloud) effects nearly compensating each other. Emissions of halogenated compounds, including CFCs and HCFCs, were assessed as very likely causing a net-positive ERF in the AR5. However, recent studies (Morgenstern et al., 2020; O’Connor et al., 2021; Thornhill et al., 2021b) find strong adjustments in Southern Hemisphere aerosols and clouds, such that the very likely range in the emission-based ERF for CFC + HCFCs + HFCs now also include negative values.
For methane emissions, in addition to their direct effect, there are indirect positive ERFs from methane enhancing its own lifetime, causing ozone production, enhancing stratospheric water vapour, and influencing aerosols and the lifetimes of HCFCs and HFCs (Myhre et al., 2013; O’Connor et al., 2021). The ERF from methane emissions is considerably higher than the ERF estimate resulting from its abundance change. The central estimate with the very likely range is 1.19 [0.81 to 1.58] W m–2 for the emissions-based estimatecompared with 0.54 W m–2 for the abundance-based estimate (Section 7.3.5). The abundance-based ERF estimate for methane results from contributions of its own emissions and the effects of several other compounds, some decreasing methane lifetime, notably NOx, which importantly reduce the methane abundance-based ERF. Emissions of CO and NMVOCs both indirectly contribute to a positive ERF through enhancing ozone production in the troposphere and increasing the methane lifetime. For CO and NMVOCs of fossil origin there is also a 0.07 W m–2 contribution to CO2 from their oxidation. The very likely total ERF of CO and NMVOCs emissions is estimated to be 0.44 [0.22 to 0.67] W m–2.
NOx causes a positive ERF through enhanced tropospheric ozone production and a negative ERF through enhanced OH concentrations that reduce the methane lifetime. There is also a small negative ERF contribution through the formation of nitrate aerosols, although only three of the AerChemMIP models include nitrate aerosols. The best estimate of the net ERF from changes in anthropogenic NOx emissions is –0.27 [–0.55 to 0.01] W m–2. The magnitude is somewhat greater than the AR5 estimate (–0.15 [–0.34 to +0.02] W m–2) but with a similar level of uncertainty. The difference between AR6 and AR5 estimates is possibly due to the different modeling protocols (see Supplementary Material: 6.SM.1).
Anthropogenic emissions of SO2 lead to the formation of sulphate aerosols and a negative ERF through aerosol–radiation and aerosol–cloud interactions. The emissions-based ERFaci, which was not previously considered in AR5, is now included. The estimated ERF is thus considerably more negative than the AR5 estimate with a radiative forcing of –0.4 W m–2, despite the decline of ERF due to aerosols since 2011 (Section 7.3.3.1.3, Figure 6.12a). SO2 emissions are estimated to contribute to a negative ERF of –0.94 [–1.63 to –0.25] W m–2, with –0.23 W m–2 from aerosol–radiation interactions and –0.70 W m–2 from aerosol–cloud interactions. Emissions of NH3 lead to formation of ammonium-nitrate aerosols with an estimated ERF of –0.03 W m–2.
The best estimate for the ERF due to emissions of BC is reduced from the AR5, and is now estimated to be 0.11 [–0.20 to 0.42] W m–2 with an uncertainty also including negative values. As discussed in Section 7.3.3.1.2, a significant portion of the positive BC forcing from aerosol–radiation interactions is offset by negative atmospheric adjustments due to cloud changes, as well as lapse rate and atmospheric water vapour changes, resulting in a smaller positive net ERF for BC compared with AR5. The large range in the forcing estimate stems from variation in the magnitude and sign of atmospheric adjustments across models and is related to the differences in the model treatment of different processes affecting BC (e.g., ageing, mixing) and its interactions with clouds and cryosphere (Section 7.3.3; Thornhill et al., 2021b). The emissions-based ERF for organic carbon aerosols is –0.21 [–0.44 to +0.02] W m–2, a weaker estimate compared with AR5 attributed to stronger absorption by OC (Section 7.3.3.1.2).
The emissions-based contributions to GSAT change (Figure 6.12b) were not assessed in AR5, but with the ERF from aerosol–cloud interactions attributed to the emitted compounds there is now a better foundation for this assessment. The contribution to emissions-based ERF at 2019 (Figure 6.12a) is scaled by the historical emissions (over the period 1750–2019) of each compound to reconstruct the historical time series of ERF. An impulse response function (Cross-Chapter Box 7.1, Supplementary Material 7.SM5.2) is then applied to obtain the contribution of SLCF emissions to the GSAT response. Due to the non-linear chemical and physical processes described above relating emissions to ERF, and the additional non-linear relations between ERF and GSAT, these emissions-based estimates of GSAT responses strongly depend on the methodology applied to estimate ERF and GSAT (Supplementary Material 6.SM.2). Therefore, the relative contribution of each compound through its primary emissions versus secondary formation or destruction (e.g., for methane emissions its ozone versus methane contributions), by construction (omitting the non-linear processes), will be equal for ERF and GSAT. Uncertainties in the GSAT response are estimated using the assessed range of the equilibrium climate sensitivity (ECS) from Chapter 7 of this report. For most of the emitted compounds the uncertainty in the GSAT response is dominated by the uncertainty in the relationship between emissions and the ERF.
The contributions from the emitted compounds to GSAT broadly follow their contributions to the ERF, mainly because their evolution over the past decades have been relatively similar and slow enough compared to their lifetimes to be reflected similarly in their ERF and GSAT despite the delay of the GSAT response to ERF changes (Section 6.6.1). However, for some SLCFs (e.g., SO2) that have been reduced globally, their contribution to GSAT change is slightly higher compared with that of CO2 than their relative contribution to ERF because the peak in their ERF change has already occurred (Section 6.4.1) whereas the peak of their GSAT effect started to decline recently (Figure 7.9). This is due to the inertia of the climate system delaying the full response of GSAT to a change in forcing (Figure 6.15).
In summary, emissions of SLCFs, especially methane, NOx and SO2, have substantial effects on effective radiative forcing (ERF) (high confidence). The net global emissions-based ERF of NOx is negative and that of NMVOCs is positive, in agreement with the AR5 assessment (high confidence). For methane, the emissions-based ERF is twice as high as the abundance-based ERF (high confidence). SO2 emissions make the dominant contribution to the ERF associated with the aerosol–cloud interaction (high confidence). The contributions from the emitted compounds to GSAT broadly follow their contributions to the ERF (high confidence). However, due to the inertia of the climate system delaying the full GSAT response to a change in forcing, the contribution to GSAT change due to SO2 emissions is slightly higher compared with that due to CO2 emissions (than their relative contributions to ERF) because the peak in emission-induced SO2 eRF has already occurred.
6.4.3 Climate Responses to SLCFs
This section briefly discusses the climate response to SLCFs, in particular to changes in aerosols, and gathers complementary information and assessments from Chapters 3, 7, 8 and 10.
Warming or cooling atmospheric aerosols, such as BC and sulphate, can affect temperature and precipitation in distinct ways by modifying the shortwave and longwave radiation, the lapse rate of the troposphere, and influencing cloud microphysical properties (Section 10.1.4, Box 8.1). An important distinction between scattering and absorbing aerosols is the opposing nature of their influences on circulation, clouds and precipitation, besides surface temperature as evident from the contrasting regional climate responses to regional aerosol emissions (e.g., Lewinschal et al., 2019; Sand et al., 2020; also see Chapters 8 and 10).
On the global scale, as assessed in Chapter 3, anthropogenic aerosols have likely cooled GSAT since 1850–1900 driven by the negative aerosol forcing, while it is extremely likely that human-induced stratospheric ozone depletion has primarily driven stratospheric cooling between 1979 and the mid-1990s. Multiple modelling studies support the understanding that present-day emissions of SO2, a precursor for sulphate aerosols, are the dominant driver of near- surface air temperature responses in comparison to BC or OC even though, for some regions, BC forcing plays a key role (Baker et al. , 2015; Samset et al. , 2016; Stjern et al. , 2017; Zanis et al. , 2020) . Further, there is high confidence that the aerosol-driven cooling has led to detectable large-scale water-cycle changes since at least the mid-20thcentury as assessed in Chapter 8. The overall effect of surface cooling from anthropogenic aerosols is to reduce global precipitation and alter large-scale atmospheric circulation patterns (high confidence), primarily driven by the cooling effects of sulphate aerosols (Section 8.2.1). In addition, there is high confidence that darkening of snow through the deposition of black carbon and other light-absorbing particles enhances snowmelt (Section 7.3.4.3; SROCC Chapter 3).
In AR5, there was low confidence in the overall understanding of climate response to spatially varying patterns of forcing, though there was medium to high confidence in some regional climate responses, such as the damped warming of the NH and shifting of the ITCZ from aerosols, and positive feedbacks enhancing the local response from high-latitude snow and ice albedo changes. Since AR5, the relationship between inhomogeneous forcing and climate response is better understood, providing further evidence of the climate influence of SLCFs (aerosols and ozone in particular) on global to regional scales (Collins et al., 2013; Shindell et al., 2015; Aamaas et al., 2017; Kasoar et al., 2018; Persad and Caldeira, 2018; Wilcox et al., 2019) which differ from the relatively homogeneous spatial influence from LLGHGs.
Large geographical variations in aerosol ERFs (Section 6.4.1) affect global and regional temperature responses (Myhre et al., 2013; Shindell et al., 2015). Multi-model CMIP6 ensemble-mean results (Figure 6.13) show cooling over almost all areas of the globe in response to increases of aerosol and their precursor emissions from 1850 to the recent past (1995–2014). While the ERF has hotspots, the temperature response is more evenly distributed in line with the results of CMIP5 models including the temperature response to ozone changes (Shindell et al., 2015). The ensemble-mean global mean surface temperature decreases by 0.66°C ± 0.51°C while decreasing by 0.97°C ± 0.54°C for the Northern Hemisphere and 0.34°C ± 0.2°C for the Southern Hemisphere. The zonal-mean temperature response is negative at all latitudes (high confidence) and becomes more negative with increasing latitude, with a maximum ensemble-mean decrease of around 2.7°C at northern polar latitudes. The zonal-mean response is not directly proportional to the zonal-mean forcing, especially in the Arctic where the temperature response is cooling while the local ERF is positive (Figure 6.10). This is consistent with prior studies showing that the Arctic, in particular, is highly sensitive to forcing at NH mid-latitudes (e.g., Shindell and Faluvegi, 2009; Sand et al., 2013a) and with results from CMIP5 models (more on the Arctic below; Shindell et al., 2015). Thus, there is high confidence that the temperature response to aerosols is more asymmetric than the response to WMGHGs and negative at all latitudes.
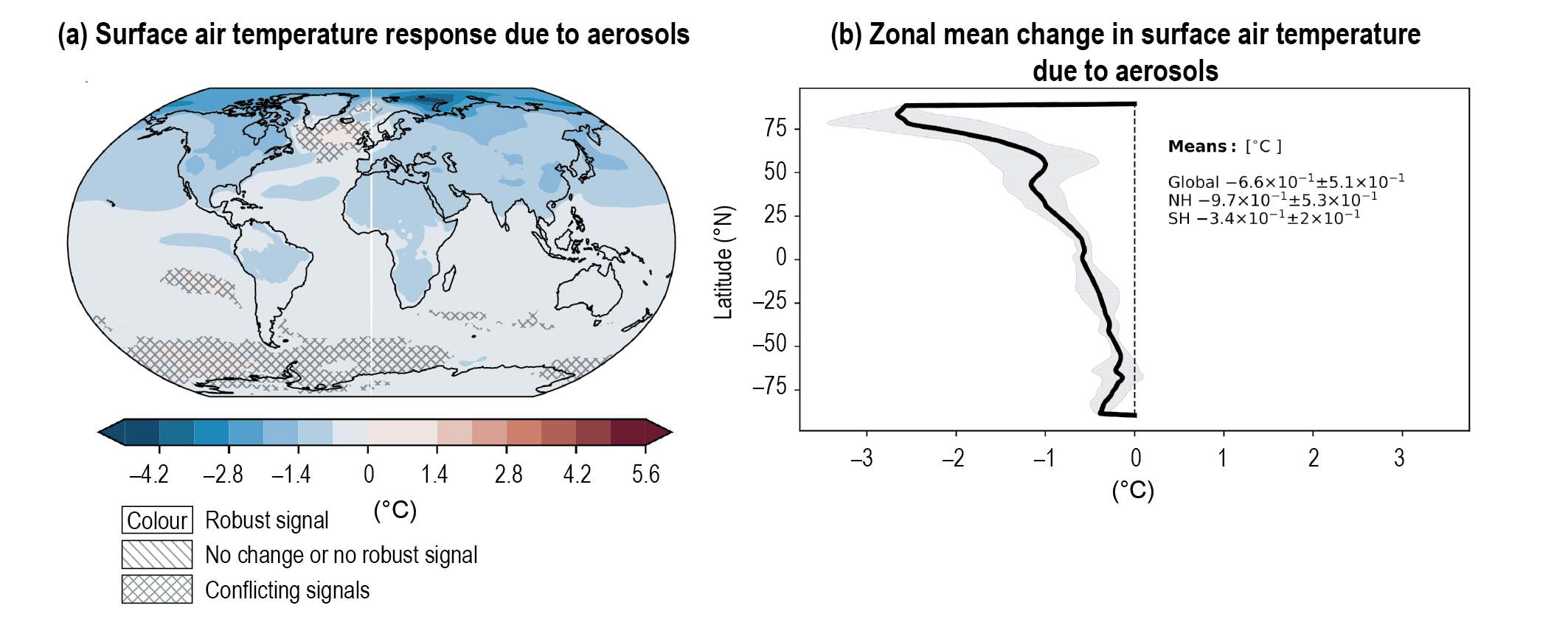 Figure 6.13 | Multi-model mean surface air temperature response over the recent past (1995–2014) induced by aerosol changes since 1850. Calculation is based on the difference between CMIP6 ‘historical’ and AerChemMIP ‘hist-piAer’ experiments averaged over 1995–2014, where (a) is the spatial pattern of the annual mean surface air temperature response, and (b) is the mean zonally averaged response. Model means are derived from the years 1995–2014. Uncertainty is represented using the advanced approach: No overlay indicates regions with robust signal, where ≥66% of models show change greater than variability threshold and ≥80% of all models agree on sign of change; diagonal lines indicate regions with no change or no robust signal, where <66% of models show a change greater than the variability threshold; crossed lines indicate regions with conflicting signal, where ≥66% of models show change greater than variability threshold and <80% of all models agree on sign of change. For more information on the advanced approach, please refer to the Cross-Chapter Box Atlas.1. AerChemMIP models MIROC6, MRI-ESM2-0, NorESM2-LM, GFDL-ESM4, GISS-E2-1-G and UKESM1-0-LL are used in the analysis. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.13 | Multi-model mean surface air temperature response over the recent past (1995–2014) induced by aerosol changes since 1850. Calculation is based on the difference between CMIP6 ‘historical’ and AerChemMIP ‘hist-piAer’ experiments averaged over 1995–2014, where (a) is the spatial pattern of the annual mean surface air temperature response, and (b) is the mean zonally averaged response. Model means are derived from the years 1995–2014. Uncertainty is represented using the advanced approach: No overlay indicates regions with robust signal, where ≥66% of models show change greater than variability threshold and ≥80% of all models agree on sign of change; diagonal lines indicate regions with no change or no robust signal, where <66% of models show a change greater than the variability threshold; crossed lines indicate regions with conflicting signal, where ≥66% of models show change greater than variability threshold and <80% of all models agree on sign of change. For more information on the advanced approach, please refer to the Cross-Chapter Box Atlas.1. AerChemMIP models MIROC6, MRI-ESM2-0, NorESM2-LM, GFDL-ESM4, GISS-E2-1-G and UKESM1-0-LL are used in the analysis. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). The asymmetric aerosol and greenhouse gas forcing on regional-scale climate responses have also been assessed to lead to contrasting effects on precipitation in Chapter 8. The asymmetric historical radiative forcing due to aerosols led to a southward shift in the tropical rain belt (high confidence) and contributed to the Sahel drought from the 1970s to the 1980s (high confidence). Furthermore, the asymmetry of the forcing led to contrasting effects in monsoon precipitation changes over West Africa, Southern Asia and Eastern Asia over much of the mid-20thcentury due to GHG-induced precipitation increases counteracted by anthropogenic aerosol-induced decreases (high confidence) (see Section 8.3 and Box 8.1).
The Arctic region is warming considerably faster than the rest of the globe (Atlas 11.2.2) and, generally, studies indicate that this amplification of the temperature response toward the Arctic has an important contribution from local and remote aerosol forcing (Stjern et al., 2017; Westervelt et al., 2018). Several studies indicate that changes in long-range transport of sulphate and BC from northern mid-latitudes can potentially explain a significant fraction of Arctic warming since the 1980s (e.g., Navarro et al. , 2016; Breider et al. , 2017; Ren et al. , 2020). Modelling studies show that changes in mid-latitude aerosols have influenced Arctic climate by changing the radiative balance through aerosol–radiation and aerosol–cloud interactions, and enhancing poleward heat transport (Navarro et al., 2016; Ren et al., 2020). Idealized aerosol-perturbation studies have shed further light on the sensitivity of Arctic temperature response to individual aerosol species. Studies show relatively large responses in the Arctic to BC perturbations and reveal the importance of remote BC forcing by rapid adjustments (Sand et al. , 2013b; Stjern et al. , 2017; L. Liu et al. , 2018; Yang et al. , 2019b). Perturbations in SO2 emissions over major emitting regions in the Northern Hemisphere have been shown to produce the largest Arctic temperature responses (Kasoar et al., 2018; Lewinschal et al., 2019).
The effects of changes in aerosols on local and remote changes in temperature, circulation and precipitation are sensitive to a number of model uncertainties affecting aerosol sources, transformation and resulting radiative efficacy. Therefore, regional climate effects in global model studies must be interpreted with caution. When investigating the climate response to regional aerosol emissions, such uncertainties are likely to be confounded even further by the variability between models in regional climate and circulation patterns, leading to greater inter-model spread at regional scales than at a global scale (Baker et al., 2015; Kasoar et al., 2016).
In summary, over the historical period, changes in aerosols and their ERF have primarily contributed to cooling, partly masking the human-induced warming (high confidence). Radiative forcings induced by aerosol changes lead to both local and remote changes in temperature (high confidence). The temperature response preserves hemispheric asymmetry of the ERF but is more latitudinally uniform with strong amplification of the temperature response towards the Arctic (medium confidence).
6.4.4 Indirect Radiative Forcing Through Effects of SLCFs on the Carbon Cycle
Deposition of reactive nitrogen (Nr; i.e., NH3 and NOx) increases the plant productivity and carbon sequestration in N-limited forests and grasslands, and also in open and coastal waters and open ocean. Such inadvertent fertilization of the biosphere can lead to eutrophication and reduction in biodiversity in terrestrial and aquatic ecosystems. The AR5 assessed that it is likely that Nr deposition over land currently increases natural CO2 sinks, in particular in forests, but the magnitude of this effect varies between regions (Ciais et al., 2013). Increasing Nr deposition or the synergy between increasing Nr deposition and atmospheric CO2 concentration could have contributed to the increasing global-net land CO2 sink (Section 5.2.1.4.1).
Ozone uptake itself damages photosynthesis and reduces plant growth with consequences for the carbon and water cycles (Ainsworth et al., 2012; Emberson et al., 2018). The AR5 concluded there wasrobust evidence of the effect of ozone on plant physiology and subsequent alteration of the carbon storage, but considered insufficient quantification of and a lack of systematic incorporation of the ozone effect in carbon-cycle models as a limitation to assess the terrestrial carbon balance (Ciais et al., 2013). Since AR5 several more ESMs have incorporated interactive ozone-vegetation damage resulting in an increase in evidence to support the influence of ozone on the land carbon cycle. The new modelling studies tend to focus on ozone effects on plant productivity rather than the land carbon storage and agree that ozone-induced gross-primary productivity (GPP) losses are largest today in the eastern USA, Europe and eastern China, ranging from 5–20% on the regional scale (low confidence) (Yue and Unger, 2014; Lombardozzi et al. , 2015; Yue et al. , 2017; Oliver et al. , 2018). There is medium evidence and high agreement based on observational studies and models that ozone-vegetation interactions further influence the climate system, including water and carbon cycles by affecting stomatal control over plant transpiration of water vapour between the leaf surface and atmosphere (Wittig et al. , 2007; Sun et al. , 2012; VanLoocke et al. , 2012; Lombardozzi et al. , 2013; Hoshika et al. , 2015; Arnold et al. , 2018). While some modelling studies suggest that the unintended Nr deposition fertilization effect in forests may potentially offset the ozone-induced carbon losses (Felzer et al., 2007; de Vries et al., 2017), complex interactions have been observed between ozone and Nr deposition to ecosystems that have not yet been included in ESMs. For some plants, the effects of increasing ozone on root biomass become more pronounced as Nr deposition increased, and the beneficial effects of Nr on root development were lost at higher ozone treatments (Mills et al., 2016). Reducing uncertainties in ozone vegetation damage effects on the carbon cycle requires improved information on the sensitivity of different plant species to ozone, and measurements of ozone dose-response relationships for tropical plants, which are currently lacking. Surface ozone effect on the land carbon sink and indirect CO2 forcing, therefore, remains uncertain. Collins et al. (2010) showed that adding in the effects of surface ozone on vegetation damage and reduced uptake of CO2 added about 10% to the methane emissions metrics and could change the sign of the NOx metrics. However, this estimate has to be considered as an upper limit due to limitation of the paramaterization used bySitch et al. (2007) considering more recent knowledge and is thus not included in the current metrics (Section 7.6.1.3).
Tropospheric aerosols influence the land and ocean ecosystem productivity and the carbon cycle through changing physical climate and meteorology (Jones, 2003; Cox et al., 2008; Mahowald, 2011; Unger et al., 2017) and through changing deposition of nutrients including nitrogen, sulphur, iron and phosphorous (Mahowald et al., 2017; Kanakidou et al., 2018). There is robust evidence and high agreement from field (Oliveira et al. , 2007; Cirino et al. , 2014; Rap et al. , 2015; X. Wang et al. , 2018) and modelling(Mercado et al., 2009; Strada and Unger, 2016; Lu et al., 2017; Yue et al., 2017) studies that aerosols affect plant productivity through increasing the diffuse fraction of downward shortwave radiation, although the magnitude and importance to the global land carbon sink is controversial. At large scales the dominant effect of aerosols on the carbon cycle is likely a global cooling effect of the climate (medium confidence) (Jones, 2003; Mahowald, 2011; Unger et al., 2017). We assess that these interactions between aerosols and the carbon cycle are currently too uncertain to constrain quantitatively the indirect CO2 forcing.
In summary, reactive nitrogen, ozone and aerosols affect terrestrial vegetation and the carbon cycle through deposition and effects on large-scale radiation (high confidence) but the magnitude of these effects on the land carbon sink, ecosystem productivity and indirect CO2 forcing remain uncertain due to the difficulty in disentangling the complex interactions between the effects. As such, we assess the effects to be of second order in comparison to the direct CO2 forcing (high confidence) but, at least for ozone, it could add a substantial (positive) forcing compared with its direct forcing (low confidence).
6.4.5 Non-CO2 biogeochemical Feedbacks
Climate change-induced changes in atmospheric composition and forcing due to perturbations in natural processes constitute an Earth system feedback amplifying (positive feedback) or diminishing (negative feedback) the initial climate perturbation (Ciais et al., 2013; Heinze et al., 2019). Quantification of these biogeochemical feedbacks is important to allow for a better estimate of the expected effects of emissions reduction policies for mitigating climate change and the effect on the allowable global carbon budget (Lowe and Bernie, 2018). Biogeochemical feedbacks due to changes in the carbon cycle are assessed in Section 5.4.5, while physical and biogeophysical climate feedbacks are assessed in Section 7.4.2. Additionally, non-CO2 biogeochemical feedbacks due to climate-driven changes in methane sources and N2O sources and sinks are assessed in Section 5.4.7. The goal of this section is to estimate the feedback parameter ( α as defined in section 7.4.1.1) from climate-induced changes in atmospheric abundances or lifetimes of SLCFs mediated by natural processes or atmospheric chemistry. These non-CO2 biogeochemical feedbacks act on time scales of years to decades and have important implications for climate sensitivity and emissions-abatement policies. The feedback parameter is quantified entirely from ESMs that expand the complexity of CCMs by coupling the physical climate and atmospheric chemistry to land and ocean biogeochemistry. In AR5, α for non-CO2 biogeochemical feedbacks was estimated from an extremely limited set of modelling studies with much less confidence associated with the estimate. Since AR5, ESMs have advanced to include more feedback processes, facilitating a relatively more robust assessment of α . CMIP6 ESMs participating in AerChemMIP performed coordinated sets of experiments (Collins et al., 2017) facilitating the consistent estimation of α (Thornhill et al., 2021a) and we rely on this multi-model analysis for the best estimates (Table 6.8). Considering the consistent methodology, the assessed central values and 5–95% ranges for α are based on the AerChemMIP estimates. The full range of model uncertainty is not captured in AerChemMIP because of the relatively small ensemble size, therefore estimates from studies using other models or with different protocols are discussed to reinforce or critique these values.
 where
where  is the change in TOA energy balance in response to a change in x induced by a change in surface temperature (T). The 5–95% range is calculated as mean ± standard deviation × 1.645 for each feedback. The level of confidence in these estimates is lowowing to the large intermodel spread. Published estimates of α x are also shown for comparison.
is the change in TOA energy balance in response to a change in x induced by a change in surface temperature (T). The 5–95% range is calculated as mean ± standard deviation × 1.645 for each feedback. The level of confidence in these estimates is lowowing to the large intermodel spread. Published estimates of α x are also shown for comparison. Non-CO2 Biogeochemical Climate Feedback (x) | Number of AerChemMIP Models | Assessed Central Estimate and Very Likely Range of Feedback Parameter ( α x) W m−2°C−1 | Published Estimates of α x W m−2°C−1 |
Sea salt | 6 | –0.049 [–0.13 to +0.03] | –0.08 (Paulot et al., 2020) |
DMS | 3 | 0.005 [0.0 to 0.01] | –0.02 (Ciais et al., 2013) |
Dust | 6 | –0.004 [–0.02 to +0.01] | –0.04 to +0.02 (Kok et al., 2018) |
Ozone | 4 | –0.064 [–0.08 to -0.04] | –0.015 (Dietmüller et al., 2014), –0.06 (Muthers et al., 2014, stratospheric ozone changes only), –0.01 (Marsh et al., 2016, stratospheric ozone changes only), –0.13 (Nowack et al., 2015, stratospheric ozone and water vapour changes), –0.007 ± 0.009 (Heinze et al., 2019, tropospheric ozone changes only) |
BVOC | 4 | –0.05 [–0.22 to +0.12] | –0.06 (Scott et al., 2017, aerosol effects only), –0.01 (Paasonen et al., 2013; indirect aerosol effects only), 0–0.06 (Ciais et al., 2013) |
Lightning | 4 | –0.010 [–0.04 to +0.02] | |
Methane lifetime | 4 | –0.030 [–0.12 to +0.06] | –0.30 ± 0.01 (Heinze et al., 2019) |
Total non-CO2 Biogeochemical feedbacks assessed in this chapter | –0.200 [–0.41 to +0.01] | 0.0 ± 0.15 (Sherwood et al., 2020) |
Climate–sea-spray feedback: Sea-spray emissions from ocean surfaces influence climate directly or indirectly through the formation of CCN as discused in Section 6.2.1.2. They are sensitive to SST and sea ice extent, as well as to wind speed, and are therefore expected to feedback on climate (Struthers et al., 2013). However, there are large uncertainties in the strength of climate feedback from sea-spray aerosols because of the diversity in the model representation of emissions (many represent sea-salt emissions only) and their functional dependence on environmental factors noted above, in situ atmospheric chemical and physical processes affecting the sea-spray lifetime, and aerosol–cloud interactions (Struthers et al., 2013; Soares et al., 2016; Nazarenko et al., 2017). Additional work is needed to identify how sea-spray and POA emissions respond to shifts in ocean biology and chemistry in response to warming, ocean acidification and changes in circulation patterns (Cochran et al. , 2017), and affect CCN and INP formation (DeMott et al., 2016). AerChemMIP models, representing only the sea-salt emissions, agree that the sea-salt-climate feedback is negative, however there is a large range in the feedback parameter indicating large uncertainties (Table 6.8).
Climate–DMS feedback: Dimethyl sulphide (DMS) is produced by marine phytoplankton and is emitted to the atmosphere where it can lead to the subsequent formation of sulphate aerosol and CCN (Section 6.2.2.5). Changes in DMS emissions from ocean could feedback on climate through their response to changes in temperature, solar radiation, ocean mixed-layer depth, sea ice extent, wind speed, nutrient recycling or shifts in marine ecosystems due to ocean acidification and climate change, or atmospheric processing of DMS into CCN (Heinze et al., 2019). Models with varying degrees of representation of the relevant biogeochemical processes and effects on DMS fluxes produce diverging estimates of changes in DMS emissions strength under climate change resulting in large uncertainties in the DMS–sulphate–cloud albedo feedback (Bopp et al., 2004; Kloster et al., 2007; Gabric et al., 2013). In AR5, the climate-DMS feedback parameter was estimated to be –0.02 W m–2°C–1based on a single model. Since AR5, new modelling studies using empirical relationships between pH and total DMS production find that global DMS emissions decrease due to combined ocean acidification and climate change, leading to a strong positive climate feedback (Six et al., 2013; Schwinger et al., 2017). However, another study argues for a much weaker positive feedback globally due to complex and compensating regional changes in marine ecosystems (S. Wang et al., 2018). The AerChemMIP multi-model analysis suggests small positive feedback (Table 6.8), consistent with these recent studies, but with large uncertainties in the magnitude of α .
Climate–dust feedback: Mineral dust is the most abundant aerosol type in the atmosphere, when considering aerosol mass, and affects the climate system by interacting with both longwave and shortwave radiation as well as contributing to the formation of CCN and INP. Because dust emissions are sensitive to climate variability (e.g., through changes in the extent of arid land; Section 6.2.2.4), it has been hypothesized that the climate-dust feedback could be an important feedback loop in the climate system. Since AR5, an improved understanding of the shortwave absorption properties of dust as well as a consensus that dust particles are larger than previously thought has led to a revised understanding that the magnitude of radiative forcing due to mineral dust is small (Kok et al., 2017; Ryder et al., 2018). A recent study notes that global models underestimate the amount of coarse dust in the atmosphere and accounting for this limitation raises the possibility that dust emissions warm the climate system (Adebiyi and Kok, 2020). Model predictions of dust emissions in response to future climate change range from an increase (Woodward et al., 2005) to a decrease (Mahowald and Luo, 2003), thus leading to high uncertainties on the sign of the climate-dust feedback. Since AR5, Kok et al. (2018) estimated the direct dust-climate feedback parameter, from changes in the dust direct radiative effect only, to be in the range –0.04 to +0.02 W m–2°C–1. The assessed central value and the 5–95% range of the climate-dust feedback parameter based on AerChemMIP ensemble (Table 6.8) is within the range of the published estimate, however both the magnitude and sign of α are model-dependent.
Climate–ozone feedback: Changes in ozone concentrations in response to projected climate change have been shown to lead to a potential climate-atmospheric chemistry feedback. Chemistry–climate models consistently project a decrease in lower tropical stratospheric ozone levels due to enhanced upwelling of ozone-poor tropospheric air associated with surface warming-driven strengthening of the Brewer-Dobson circulation (Bunzel and Schmidt, 2013). Further, models project an increase in middle and extratropical stratospheric ozone due to increased downwelling through the strengthened Brewer-Dobson circulation (Bekki et al., 2013; Dietmüller et al., 2014). These stratospheric ozone changes induce a net-negative global mean ozone radiative feedback (Dietmüller et al., 2014). Tropospheric ozone shows a range of responses to climate with models generally agreeing that warmer climate will lead to decreases in the tropical lower troposphere owing to increased water vapour, and increases in the subtropical to mid-latitude upper troposphere due to increases in lightning and stratosphere-to-troposphere transport (Stevenson et al., 2013). A small positive feedback is estimated from climate-induced changes in global mean tropospheric ozone (Dietmüller et al., 2014) while a small negative feedback is estimated by Heinze et al. (2019) based on the model results of Stevenson et al. (2013). Additionally, these ozone feedbacks induce a change in stratospheric water vapour amplifying the feedback due to stratospheric ozone (Stuber et al., 2001). Since AR5, several modelling studies have estimated the intensity of meteorology-driven ozone feedbacks on climate from either combined tropospheric and stratospheric ozone changes or separately with contrasting results. One study suggests no change (Marsh et al., 2016), while other studies report reductions of ECS ranging from 7–8% (Dietmüller et al., 2014; Muthers et al., 2014) to 20% (Nowack et al., 2015). The estimate of this climate-ozone feedback parameter is very strongly model-dependent with values ranging from –0.13 to –0.01 W m–2°C–1though there is agreement that it is negative. The assessed central value and the 5–95% range of climate-ozone feedback parameter based on AerChemMIP ensemble is within the range of these published estimates, but closer to the lower bound. This climate-ozone feedback factor does not include the feedback on ozone from lightning changes which is discussed separately below.
Climate–BVOC feedback: BVOCs, such as isoprene and terpenes, are produced by land vegetation and marine plankton (Sections 6.2.2.3 and 6.2.2.5). Once in the atmosphere, BVOCs and their oxidation products lead to the formation of secondary organic aerosols (SOA) exerting a negative forcing, and increased ozone concentrations and methane lifetime exerting a positive forcing. BVOC emissions are suggested to lead to a climate feedback in part because of their strong temperature dependence observed under present-day conditions (Kulmala et al., 2004; Arneth et al., 2010a). Their response to future changes in climate and CO2 levels remains uncertain (Section 6.2.2.3). Estimates of the climate-BVOC feedback parameter are typically based on global models which vary in their level of complexity of emissions parametrization, BVOC speciation, the mechanism of SOA formation and the interaction with ozone chemistry (Thornhill et al., 2021a). Since AR5, observational studies (Paasonen et al., 2013) and models (Scott et al., 2018) estimate the feedback due to biogenic SOA (via changes in BVOC emissions) to be in the range of about –0.06 to –0.01 W m–2°C–1. The assessed central estimate of the climate-BVOC feedback parameter based on the AerChemMIP ensemble suggests that climate-induced increases in SOA from BVOCs will lead to a strong cooling effect that will outweigh the warming from increased ozone and methane lifetime, however the uncertainty is large (Thornhill et al., 2021a).
Climate–lightning NOxfeedback: As discussed in Section 6.2.2.1, climate change influences lightning NOx emissions. Increases in lightning NOx emissions will not only increase tropospheric ozone and decrease methane lifetime but also increase the formation of sulphate and nitrate aerosols, via oxidant changes, offsetting the positive forcing from ozone. The response of lightning NOx to climate change remains uncertain and is highly dependent on the parametrization of lightning in ESMs (Section 6.2.1.2; Finney et al., 2016b; Clark et al., 2017). AerChemMIP multi-model ensemble mean estimate a net-negative climate feedback from increases in lightning NOx in a warming world (Thornhill et al., 2021a). All AerChemMIP models use a cloud-top height lightning parametrization that predicts increases in lightning with warming. However, a positive climate-lightning NOx feedback cannot be ruled out because of the dependence of the response to lightning parametrizations as discussed in Section 6.2.2.1.
Climate–methane lifetime feedback: Warmer and wetter climate will lead to increases in OH and oxidation rates leading to reduced atmospheric methane lifetime – a negative feedback (Naik et al., 2013; Voulgarakis et al., 2013). Furthermore, since OH is in turn removed by methane, the climate-methane lifetime feedback will be amplified (Section 6.3.1; Prather, 1996). Based on the multi-model results of Voulgarakis et al. (2013), α for climate-methane lifetime is estimated to be –0.030 ± 0.01 W m−2°C−1by Heinze et al. (2019). The assessed central value of α based on the AerChemMIP ensemble is within the range of this estimate but with greater uncertainty (Thornhill et al., 2021a).
Climate–fire feedback: Wildfires are a major source of SLCF emissions (Section 6.2.2.6). Climate change has the potential to enhance fire activity (Sections 12.4 and 5.4.3.2) thereby enhancing SLCF emissions leading to feedbacks. Climate-driven increases in fire could potentially lead to offsetting feedback from increased ozone and decreased methane lifetime (due to increases in OH) leaving the feedback from aerosols to dominate with an uncertain net effect (e.g., Landry et al., 2015). The AR5 assessment of climate-fire feedbacks included a value of α due to fire aerosols to be in the range of –0.03 to + 0.06 W m−2°C−1based on Arneth et al. (2010a) . A recent study estimates climate feedback due to fire aerosols to be greater than that due to BVOCs, with a value of α equal to –0.15 (–0.24 to –0.05) W m−2°C−1 (Scott et al., 2018). Clearly, the assessment of fire-related non-CO2 biogeochemical feedbacks is very uncertain because of limitations in the process understanding of the interactions between climate, vegetation and fire dynamics, and atmospheric chemistry and their representation in the current generation ESMs. Some AerChemMIP ESMs include the representation of fire dynamics but do not activate their interaction with atmospheric chemistry. Given the large uncertainty and lack of information from AerChemMIP ESMs, we do not include a quantitative assessment of climate-fire feedback for AR6.
In summary, climate-driven changes in emissions, atmospheric abundances or lifetimes of SLCFs are assessed to have anoverall cooling effect, that is, a negative feedback parameter of –0.20 [–0.41 to + 0.01] W m−2°C−1, thereby reducing climate sensitivity (Section 7.4.2.5.1). This net feedback parameter is obtained by summing the assessed estimates for the individual feedback given in Table 6.8. confidence in the magnitude and the sign of most of the individual as well as the total non-CO2 biogeochemical feedbacks remains low as evident from the large range in the value of α . This large uncertainty is attributed to the diversity in model representation of the relevant chemical and biogeochemical processes based on limited process-level understanding.
6.4.6 ERF by Aerosols in Proposed Solar Radiation Modification
Solar radiation modification (SRM; Sections 4.6.3.3 and 8.6.3) has the potential to exert a significant ERF on the climate, mainly by affecting the SW component of the radiation budget (e.g., Caldeira et al., 2013; NRC, 2015; Lawrence et al., 2018). The possible ways and the extent to which the most commonly discussed options may affect radiative forcing is addressed in this section. Side effects of SRM on stratospheric ozone and changes in atmospheric transport due to radiative heating of the lower stratosphere are discussed in Section 4.6.3.3.
Stratospheric aerosol injections (SAI) have the potential to achieve a high negative global ERF, with maximum ERFs ranging from –5 to –2 W m–2 (Niemeier and Timmreck, 2015; Weisenstein et al., 2015; Niemeier and Schmidt, 2017; Kleinschmitt et al., 2018). The magnitude of the maximum achievable ERF depends on the chosen aerosol type and mixture, internal structure and size, or precursor gas (e.g., SO2), as well as the injection strategy (latitude, altitude, magnitude and season of injections), plume dispersal, model representation of aerosol microphysics, and ambient aerosol concentrations (Rasch et al. , 2008; Robock et al. , 2008; Pierce et al. , 2010; Weisenstein et al. , 2015; Laakso et al. , 2017; MacMartin et al. , 2017; Dai et al. , 2018; Kleinschmitt et al. , 2018; Vattioni et al. , 2019; Visioni et al. , 2019). For sulphur, the radiative forcing efficiency is of around –0.1 to –0.4 W m–2/(TgS yr–1) (Niemeier and Timmreck, 2015; Weisenstein et al., 2015; Niemeier and Schmidt, 2017). Different manufactured aerosols, such as ZrO2, TiO2 and Al2O3, have different ERF efficiencies compared to sulphate (Ferraro et al. , 2011; Weisenstein et al. , 2015; Dykema et al. , 2016; Jones et al. , 2016). The aerosol size distribution influences the optical properties of an aerosol layer, and hence the ERF efficiency, which also depends on the dispersion, transport, and residence time of the aerosols.
For marine cloud brightening(MCB), seeded aerosols may affect both cloud microphysical and macrophysical properties (Section 7.3.3.2). By principle, MCB relies on ERFaci through the so-called Twomey effect (Twomey, 1977), but ERFari may be of equal magnitude as shown in studies that consider spraying of sea salt outside tropical marine cloud areas (Jones and Haywood, 2012; Partanen et al., 2012; Alterskjaer and Kristjánsson, 2013; Ahlm et al., 2017). The maximum negative ERF estimated from modelling is within the range of –5.4 to –0.8 W m–2 (Latham et al. , 2008; Rasch et al. , 2009; Jones et al. , 2011; Partanen et al. , 2012; Alterskjaer and Kristjánsson, 2013). For dry sea salt, the ERF efficiency is estimated to be within the range of –3 to –10 W m–2/(Pg yr–1), when emitted over tropical oceans in ESMs in the Geoengineering Intercomparison Project (GeoMIP; Ahlm et al., 2017). Cloud-resolving models reveal the complex behaviour and response of stratocumulus clouds to seeding, in that the ERF efficiency depends on meteorological conditions, and the ambient aerosol composition, where lower background particle concentrations may increase the ERFaci efficiency (Wang et al., 2011). Seeding could suppress precipitation formation and drizzle, and hence increase the lifetime of clouds, preserving their cooling effect (Ferek et al., 2000). In contrast, cloud lifetime could be decreased by making the smaller droplets more susceptible to evaporation. Modelling studies have shown that a positive ERFaci (warming) could also result from seeding clouds with too large aerosols (Pringle et al., 2012; Alterskjaer and Kristjánsson, 2013). These individual and combined processes are not well understood, and may have a limited representation in models, or counteracting errors (Mülmenstädt and Feingold, 2018), lending low to medium confidence to the ERF estimates.
Modelled ERFaci associated with cirrus cloud thinning (CCT) cover a wide range in the literature, and the maximum are of the order of –0.8 to –3.5 W m–2, though they are of low confidence, with some studies using more simplified representations (Mitchell and Finnegan, 2009; Storelvmo et al., 2013; Kristjánsson et al., 2015; Jackson et al., 2016; Muri et al., 2018; Gasparini et al., 2020). ERFaci for CCT is mainly affected by particle seeding concentrations, with an optimum around 20 L–1, according tolimited evidence from models (Storelvmo et al., 2013). Seeding leading to higher particle concentrations could lead to a warming (Storelvmo et al., 2013; Penner et al., 2015; Gasparini and Lohmann, 2016). The lack of representation of processes related to, for example, heterogeneous and homogeneous freezing and their prevalence, is a dominant source of uncertainty in ERF estimates, in addition to less research activity.
In summary, the aerosol and cloud microphysics involved with SRM are not well understood, notably due to insufficient (and varying degrees of) representation of relevant processes in models. ERF of up to several W m–2 is reported in the literature, with SAI at the higher end and CCT with lower potentials, though it remains a challenge to establish ERF potentials and efficacies with confidence. Modelling studies have been published with more sophisticated treatment of SRM since AR5, but the uncertainties, such as cloud–aerosol radiation interactions, remain large (high confidence).
6.5 Implications of Changing Climate on AQ
Air pollutants can be impacted by climate change through physical changes affecting meterorological conditions, chemical changes affecting their lifetimes, and biological changes affecting their natural emissions (Kirtman et al., 2013). Changes in meteorology affect air quality directly through modifications of atmospheric transport patterns (e.g., occurrence and length of atmospheric blocking episodes, ventilation of the polluted boundary layer), extent of mixing layer and stratosphere–troposphere exchange (STE) for surface ozone (von Schneidemesser et al., 2015), and through modifications of the rate of reactions that generate secondary species in the atmosphere. Changing precipitation patterns in a future climate also influence the wet removal efficiency, in particular for atmospheric aerosols (Hou et al., 2018). Processes at play in non-CO2 biogeochemical feedbacks (Section 6.4.5) are also involved in the perturbation of atmospheric pollutants (Section 6.2.2).
This section relies on observational studies performed by analysing the correlation between specific meteorological conditions projected to occur more frequently in the future and surface pollutants, and global- and regional-scale modelling studies considering solely climate change in the future. We also assess the surface ozone and PM2.5 changes based on CMIP6 models analysed in AerChemMIP, considering climate change in isolation with emissions in 2050 from SSP3-7.0 scenario (Section 6.7.1). Air quality being highly variable in space and time, the use of regional atmospheric chemistry models is necessary to characterize the effect of future climate on air quality properly. However, difficulties for such assessment arise from the need for long simulations that include complex chemistry–natural system interactions with high computational cost, in addition to the difficulty related to the regionalization of climate change (Section 10.3.1.2). Changes in the occurrence of weather patterns influencing air pollution (e.g., anticyclonic stagnation conditions, transport pathways from pollution sources, convection) due to climate change are assessed in Chapters 4 and 11.
6.5.1 Effect of Climate Change on Surface Ozone
The AR5 assessed with high confidence that in unpolluted regions, higher water vapour abundances and temperatures in a warmer climate would enhance ozone chemical destruction, leading to lower baseline5surface ozone levels (Kirtman et al., 2013). In polluted regions, AR5 assessed with medium confidence that higher surface temperatures will trigger regional feedbacks in chemistry and local emissions that will increase surface ozone and the intensity of surface ozone peaks.
The response of surface ozone to climate-induced Earth system changes is complex due to counteracting effects. Studies considering the individual effects of climate-driven changes in specific precursor emissions or processes show increases in surface ozone under warmer atmosphere for some processes. This is indeed the case for enhanced STE and stratospheric ozone recovery (Sekiya and Sudo, 2014; Hess et al., 2015; Banerjee et al., 2016; Meul et al., 2018; Morgenstern et al., 2018; Akritidis et al., 2019) or the increase of soil NOx emissions (Wu et al., 2008; Romer et al., 2018), which can each lead to 1 to 2 ppb increase in surface ozone. Other processes, in particular deposition or those related to emissions from natural systems (Section 6.2.2) are expected to play a key role in future surface ozone and even the occurrence of pollution events (e.g., in the case of wildfires) but their effects are difficult to quantify in isolation.
Since the AR5, several studies have investigated the net effect of climate change on surface ozone, based on either global or regional model projections. A systematic and quantitative comparison of the ozone change, however, is difficult due to the variety of models with different complexities in the representation of natural emissions, chemical mechanisms and physical processes, as well as the surface ozone metrics applied for analysis. Ozone response to climate change has been shown to be particularly sensitive to model representation of processes like BVOC emissions, deposition, and isoprene chemistry(Squire et al. , 2015; Val Martin et al. , 2015; Schnell et al. , 2016; Pommier et al. , 2018) . More robust protocols are now used more commonly comprising, notably, longer simulations necessary to separate change from interannual variability (Barnes et al. , 2016; Lacressonnière et al. , 2016; Garcia-Menendez et al. , 2017). However, the amplitude of climate change penalty on ozone over polluted regions may be different in high-resolution (regional- and urban-scale) models in comparison to coarse-resolution global models, because a number of controlling processes are resolution-dependent including for example, local emissions and sensitivity to the chemical regime (NMVOC limited versus NOx limited; Lauwaet et al., 2014; Markakis et al., 2014, 2016).
Consistent with AR5 findings, global mean surface ozone concentration decreases range from 0.69 ± 0.16 ppb to 2.28 ± 0.24 ppb due to the dominating role of ozone destruction by water vapour in a four-member ensemble of CMIP6 ESM for surface warmings of 1.5°C–2.5°C (Figure 6.14). This decrease is driven by the ozone decrease over oceans, especially in the tropics (decrease of 1–5 ppb) and large parts of the continental unpolluted regions. The sensitivity of annual mean surface ozone to the level of surface warming over these remote areas varies spatially from –2 to –0.2 ppb oC–1 (Supplementary Material Figure 6.SM.1).
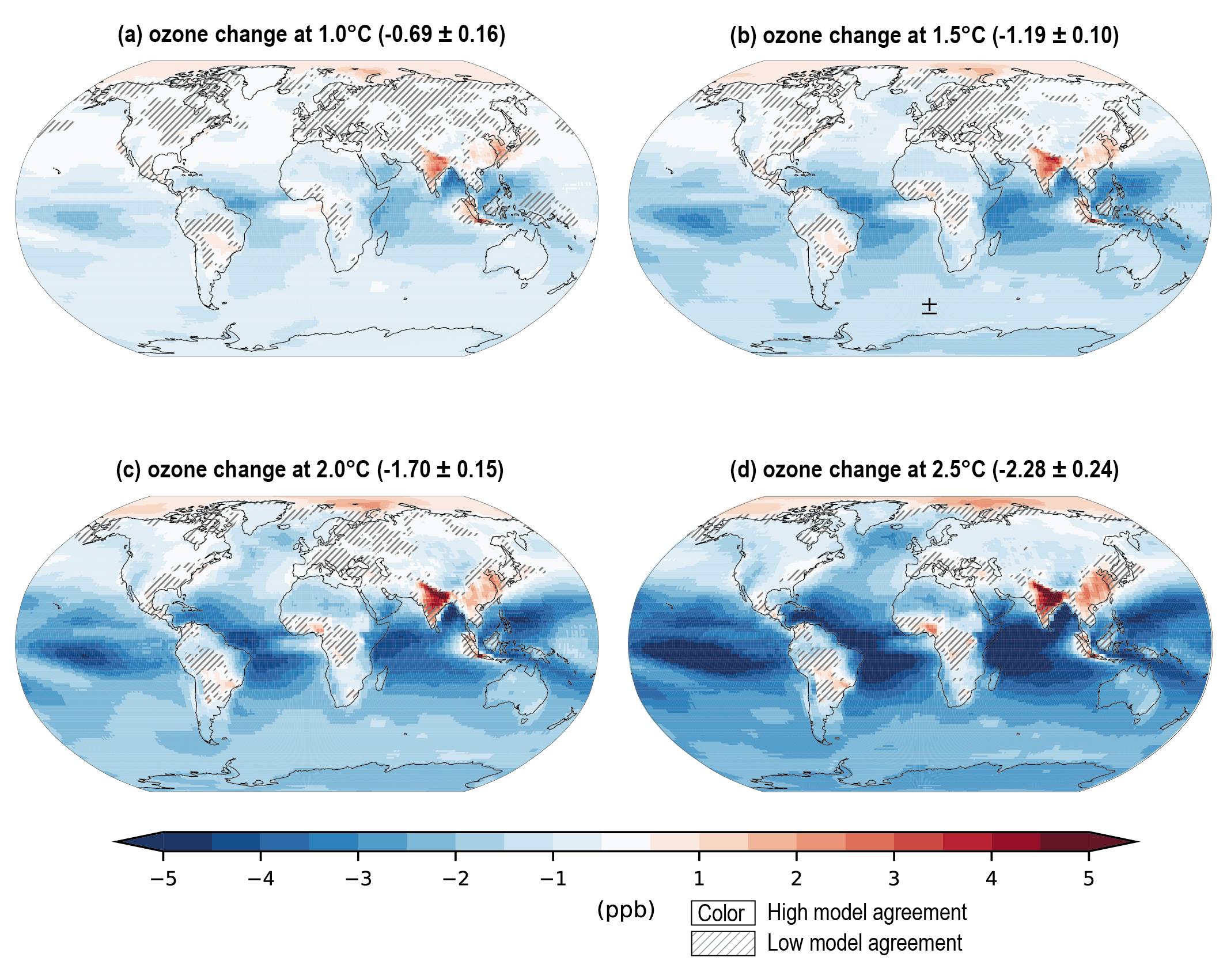 Figure 6.14 | Multi-model annual mean change in surface O3 (ppb) concentrations at different warming levels. Changes are shown for (a) 1.0°C, (b) 1.5°C, (c) 2.0°C and (d) 2.5°C increases in global mean surface air temperature. CMIP6 models include GFDL-ESM4, GISS-E2-1-G, MRI-ESM2-0 and UKESM1-0-LL. For each model, the change in surface O3 is calculated as the difference between two AerChemMIP experiments – one with evolving future emissions and sea surface temperatures (SSTs) under the SSP3-7.0 scenario and the other with the same setup but with fixed present-day SSTs. The difference is calculated as a 20-year mean in surface O3 around the year when the temperature threshold in each model is exceeded. The multi-model change in global annual mean surface O3 concentrations with ± 1standard deviation are shown within parentheses. Uncertainty is represented using the simple approach: no overlay indicates regions with high model agreement, three out of four models agree on sign of change; diagonal lines indicate regions with low model agreement, where three out of four models agree on sign of change. For more information on the simple approach, please refer to the Cross-Chapter Box Atlas.1. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.14 | Multi-model annual mean change in surface O3 (ppb) concentrations at different warming levels. Changes are shown for (a) 1.0°C, (b) 1.5°C, (c) 2.0°C and (d) 2.5°C increases in global mean surface air temperature. CMIP6 models include GFDL-ESM4, GISS-E2-1-G, MRI-ESM2-0 and UKESM1-0-LL. For each model, the change in surface O3 is calculated as the difference between two AerChemMIP experiments – one with evolving future emissions and sea surface temperatures (SSTs) under the SSP3-7.0 scenario and the other with the same setup but with fixed present-day SSTs. The difference is calculated as a 20-year mean in surface O3 around the year when the temperature threshold in each model is exceeded. The multi-model change in global annual mean surface O3 concentrations with ± 1standard deviation are shown within parentheses. Uncertainty is represented using the simple approach: no overlay indicates regions with high model agreement, three out of four models agree on sign of change; diagonal lines indicate regions with low model agreement, where three out of four models agree on sign of change. For more information on the simple approach, please refer to the Cross-Chapter Box Atlas.1. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). Over ozone-producing regions of the world, such as in North America, Europe and Eastern Asia, AR5 and post-AR5 model studies project a general increase of surface ozone levels (climate change penalty on ozone) in a future warmer climate particularly during summer(Fu and Tian, 2019). However, in current regional models, using more robust protocols, this increase of surface ozone, attributable to climate change, is of lower magnitude than in previous estimates (Lacressonnière et al., 2016; Garcia-Menendez et al., 2017). Climate change enhances the efficiency of precursor emissions to generate surface ozone in polluted regions (Schnellet al. , 2016), and thus the magnitude of this effect will depend on the emissions considered in the study (present or future, and mitigated or not; Coletteet al. , 2015; Fioreet al. , 2015).
Considering anthropogenic emissions of precursors globally higher than the current emissions (SSP3-7.0 in 2050; Figure 6.20), the CMIP6 ensemble confirms thesurface ozone penalty due to climate changeover regions close to anthropogenic pollution sources or close to natural emissions sources of ozone precursors (e.g., biomass-burning areas), with a penalty of a few ppb for the annual mean, proportional to warming levels (Figure 6.14). This rate ranges regionally from 0.2–2 ppb °C–1 (Supplementary Material Figure 6.SM.1). The CMIP6 ESMs show this consistently for South East Asia (in line with Hong et al. (2019) and Schnell et al. (2016)) and for India (in line with Pommier et al., 2018) as well as in parts of Africa and South America, close to enhanced BVOC emissions (at least three out of four ESMs agree on the sign of change). The results are mixed in polluted regions of Europe and US because of lower anthropogenic precursor emissions which leads to a very low sensitivity of surface ozone to climate change (–0.5 ppb °C–1 to 0.5 ppb °C–1; Supplementary Material Figure 6.SM.1) and thus the ESMs can disagree on sign of changes for a given warming level. This heterogeneity in the results is also found in regional studies over North America (Gonzalez-Abraham et al. , 2015; Val Martin et al. , 2015; Schnell et al. , 2016; He et al. , 2018; Nolte et al. , 2018; Rieder et al. , 2018) or over Europe (Colette et al. , 2015; Lacressonnière et al. , 2016; Schnell et al. , 2016; Fortems-Cheiney et al. , 2017).
Overall, warmer climate is expected to reduce surface ozone in unpolluted regions as a result of greater water vapour abundance accelerating ozone chemical loss (high confidence). Over regions with high anthropogenic and/or natural ozone precursor emissions, there is prevailing evidence that climate change will introduce a surface O3 penalty increasing with increasing warming levels (with a magnitude ranging regionally from 0.2–2 ppb °C–1) (medium to high confidence). Yet, there are uncertainties in processes affected in a warmer climate which can impact and modify future baseline and regional/local surface ozone levels. The response of surface ozone to future climate change through stratosphere–troposphere exchange, soil NOx emissions and wildfires is positive (medium confidence). In addition, there is low confidence in the magnitude of the effect of climate change on surface ozone through biosphere interactions (natural methane, non-methane BVOC emissions and ozone deposition) and lightning NOx emissions.
6.5.2 Impact of Climate Change on Particulate Matter
Changes in concentration and chemistry of particulate matter (PM) in a changing climate depend in a complex manner on the response of the multiple interactions of changes in emissions, chemical processes, deposition and other factors (e.g., temperature, precipitation, circulation patterns). These changes are difficult to assess and, at the time of AR5, no confidence level was attached to the overall impact of climate change on PM2.5 (Kirtman et al., 2013). Possible changes induced by climate change may concern both atmospheric concentration levels and chemical composition.
Higher temperatures increase the reaction rate of gaseous SO2 to particulate sulphate conversion but also favour evaporation of particulate ammonium nitrate (Megaritis et al., 2013). Also, higher temperatures are expected to affect BVOC emissions (e.g., Pacifico et al., 2012) that would influence SOA concentrations, although this effect has been questioned by more recent evidence (B. Wang et al., 2018; Z. Zhao et al., 2019). More generally, climate change will also affect dust concentration levels in the atmosphere (Section 6.2.2.4) and the occurrence of forest fires, both very large sources of aerosols to the global troposphere (Section 6.2.2.6).
Wet deposition constitutes the main sink for atmospheric PM (Allen et al., 2016, 2019; Xu and Lamarque, 2018). In particular, precipitation frequency has a higher effect on PM wet deposition than precipitation intensity (Hou et al., 2018). PM is also sensitive to wind speed and atmospheric stability conditions emphasizing the importance of stagnation episodes and low planetary boundary layer heights for increasing PM atmospheric concentrations (Porter et al., 2015).
At the global scale, depending on its magnitude, the warming leads either to a small increase in global mean PM concentration levels (about 0.21 µg m–3 in 2100 for RCP8.5), mainly controlled by sulphate and organic aerosols or a small decrease (–0.06 µg m–3 for RCP2.6, Westervelt et al. (2016) and Xu and Lamarque (2018)). On the other hand, Xu and Lamarque (2018) and Allen et al. (2016, 2019) found an increase of aerosol burden and PM surface concentration throughout the 21st Century, attributed to a decrease in wet-removal flux despite the overall projected increase in global precipitation, on the ground of an expected shift of future precipitation towards more frequent heavy events. Based only on three models, the CMIP6 ensembleshows that for most land areas, there is low agreement between models on the sign of the effect of climate change on annual mean PM2.5 (Supplementary Material Figure 6.SM.2).
Due to the typical atmospheric lifetime of PM in the atmosphere, of the order of a few days, most studies dealing with the future PM concentration levels have a regional character and concern mainly Europe (Megaritis et al. , 2013; Lacressonnière et al. , 2016, 2017; Lemaire et al. , 2016; Cholakian et al. , 2019), the USA (Penrod et al. , 2014; Fiore et al. , 2015; Gonzalez-Abraham et al. , 2015; Shen et al. , 2017; He et al. , 2018; Nolte et al. , 2018), Southern and Eastern Asia(Jiang et al., 2013; Nguyen et al., 2019) and India (Pommier et al., 2018). No studies are available for other areas of the world.
Changes in the chemical composition of PM as a result of future climate change can also be an important issue for the effects of PM on human health and the environment, but only sparse data are available in the literature on this and the results are, as yet, inconclusive (Im et al. , 2012; Jiang et al. , 2013; Megaritis et al. , 2013; Gonzalez-Abraham et al. , 2015; Gao et al. , 2018; He et al. , 2018; Cholakian et al. , 2019).
Overall, there is medium confidence (medium evidence, high agreement) in a small effect, positive or negative, on PM global burden due to climate change.
6.5.3 Impact of Climate Change on Extreme Pollution
Extreme air pollution is identified as the concentration of an air pollutant that is above a given threshhold value (high concentration or a high percentile) as the sensitivity of peak values to meteorological conditions can be different from sensitivity of the median or mean (Porter et al., 2015). The AR5 assessed with medium confidence that uniformly higher temperatures in polluted environments will trigger regional feedbacks in chemistry and local emissions that will increase peak ozone and PM pollution, but assessed low confidence in projecting changes in meteorological blocking associated with these extreme episodes.
Meteorological conditions, such as heatwaves, temperature inversions and atmospheric stagnation episodes favour air quality extremes and are influenced by changing climate (Fiore et al., 2015). The body of literature on the connection between climate change and extreme anthropogenic pollution episodes is essentially based on correlation and regression applied to observation reanalysis but the metrics and methodologies differ making quantitative comparisons difficult. Many emission processes in the natural systems are sensitive to temperature, and bursts of emissions as a reponse to extreme weather, as in the case of wildfires in dry conditions (Bondur et al., 2020; Xie et al., 2020) can occur, which would then add to the risk of extreme air pollution but are not sufficiently constrained to be quantitatively assessed.
Since AR5, published studies provide augmented evidence for the connections between extreme ozone and PM pollution events and high temperatures, especially long-lasting heatwaves, whose frequency is increasing due to a warming climate (Lelieveld et al., 2014; Porter et al., 2015; Hou and Wu, 2016; Jing et al., 2017; Schnell and Prather, 2017; Sun et al., 2017; H. Zhang et al., 2017). However, the relationship between air pollution and individual meteorological parameters is exaggerated because of covariation on synoptic time scales (Fiore et al., 2015). For example, heatwaves are often associated with clear skies and stagnation, making clear attribution to specific meteorological variables complicated. In Asia, future changes in winter conditions have also been shown to favour more particulate pollution (Cai et al., 2017; Zou et al., 2017). The relationship between the occurrence of stagnation episodes and high concentrations of ozone and PM2.5 has been shown to be regionally and metric dependant (Oswald et al., 2015; Sun et al., 2017; Kerr and Waugh, 2018; Schnell et al., 2018; Garrido-Perez et al., 2019).
The increase of frequency, duration and intensity of heatwaves is extremely likely on all continents for different future warming levels (Section 11.3.5, Table 11.2). However, there is low confidence in projected changes in storm tracks, jets and blocking, and thus their influence on extreme temperatures in the mid-latitudes (Section 11.3.1).
In conclusion, there is still medium confidence that climate-driven changes in meterorological conditions, such as heatwaves or stagnations, will favour extreme air pollution episodes over highly polluted areas, however, the relationship between these meteorological conditions and high concentrations of ozone and PM2.5 have been shown to be regionally and metric dependant.
6.6 Air Quality and Climate Response to SLCF Mitigation
Long-lived greenhouse gas (LLGHG) emissions reductions are typically motivated by climate change mitigation policies, whereas SLCF reductions mostly result from air pollution control and climate policies (FAQ6.2), as well as policies focusing on achieving UN Sustainable Development Goals (SDGs; Box 6.2). The management of several SLCFs (BC, methane, tropospheric ozone and HFCs) is considered in the literature as a fast-response, near-term measure to curb climate change, while reduction of emissions of LLGHGs is an essential measure for mitigating long-term climate warming (Shindell et al., 2012, 2017b; Shoemaker et al., 2013; Rogelj et al., 2014b; Lelieveld et al., 2019). Note that the term short-lived climate pollutants (SLCPs), referring only to warming SLCFs, has been used within the policy arena. The SR1.5 report states that limiting warming to 1.5°C to achieve Paris Agreement goals, implies net-zero CO2 emissions around 2050 and concurrent deep reductions in emissions of non-CO2 forcers, particularly methane (Rogelj et al., 2018a). In addition, several SLCFs are key air pollutants or precursors of fine particulate matter (PM2.5) and tropospheric ozone, and therefore subject to control driven by air-quality targets.
Policies addressing the reduction of either SLCFs or LLGHGs, often prioritize mitigation of emissions from specific anthropogenic sources, such as energy production, industry, transportation, agriculture, waste management and residential fuel use. The choice of the targetted sector and chosen measures will determine the ratios of emitted SLCFs and LLGHGs. These changes in emissions of co-emitted species will result in diverse responses driven by complex chemical and physical processes, and resulting climate perturbations. The understanding of the co-benefits through sectoral mitigation efforts (as well as potential negative impacts) is essential to inform policymaking.
The discussion of targeted SLCF policies and their role in climate change mitigation includes: critical evaluation of the climate co-benefits (Smith and Mizrahi, 2013; Pierrehumbert, 2014; Rogelj et al., 2014b; Strefler et al., 2014; M.R. Allen et al., 2016); modelling the potential of dedicated BC and methane reductions in association with or without climate policy (Harmsen et al., 2020a; Smith et al., 2020); quantifying individual or multi-component mitigation in relation to natural variability (Samset et al., 2020); warning about the risk of diversion of resources from targeted LLGHG policies, especially those targeting CO2(e.g., Shoemaker et al., 2013); and seeing it as an opportunity to strengthen commitment and accelerate action on LLGHGs (Victor et al., 2015; Aakre et al., 2018).
Over the last decade, research on air quality–climate interactions and feedbacks has brought new attention from policy communities to the possibility of win-win mitigation policies that could both improve air quality and mitigate climate change, possibly also reducing the cost of interventions (Anenberg et al. , 2012; Shindell et al. , 2012, 2017b; Schmale et al. , 2014a, b; Sadiq et al. , 2017; Fay et al. , 2018; Harmsen et al. , 2020b). Haines et al. (2017) and Shindell et al. (2017b) connect the measures to mitigate SLCFs with the achievements of some of the SDGs. Indeed, most studies on co-benefits to date focus on the impacts of climate change mitigation strategies, in particular to meet Nationally Determined Contributions (NDCs) and/or to remain below specific global temperature targets, on air quality and human health (West et al. , 2013; Rao et al. , 2016; Shindell et al. , 2016, 2017b; Zhang et al. , 2016; Chang et al. , 2017; Haines et al. , 2017; Li et al. , 2018; Markandya et al. , 2018; Williams et al. , 2018; Xie et al. , 2018; Lelieveld et al. ). Such co-benefits of climate change mitigation for air quality and human health can offset the costs of the climate measures (Saari et al., 2015; Li et al., 2018). A growing number of studies analyse the co-benefits of current and planned air-quality policies on LLGHGs and global and regional climate change impacts (Lund et al. , 2014; Akimoto et al. , 2015; Lee et al. , 2016; Maione et al. , 2016; Peng et al. , 2017).
This section assesses the effects of mitigating SLCFs, motivated by various objectives, discussing temperature response time, temperature and air-quality attribution of SLCFs sources, and chosen mitigation approach. The air quality and climate effects of the measures to contain the spread of COVID-19 in 2020 are discussed in Cross-Chapter Box 6.1 at the end of this section.
6.6.1 Implications of Lifetime on Temperature Response Time Horizon
The effect over time on GSAT following a mitigation effort affecting emissions of LLGHGs or SLCFs depends on the lifetimes of the LLGHGs and SLCFs, their radiative efficiencies, how fast the emissions are reduced, how long reductions last (limited time or sustained reduction), and the inertia of the climate system itself. Mitigation of SLCFs is often implemented through new legislation or technology standards for the different emissions sectors and components, implying that reductions are sustained over time.
It is often perceived that the full climatic response following mitigation of SLCFs will occur almost immediately. However, the inertia of the climate system strongly modifies the short-term and long-term response. SLCFs with lifetimes shorter than the time scales for inter-hemisheric mixing (1–2 years) can cause a more spatially heterogeneous forcing than LLGHGs and thus different regional patterns of the climate response (Section 6.4.3). The temporal response in GSAT to a radiative forcing can be quantified using linear impulse response functions (Cross-Chapter Box 7.1; Geoffroy et al. , 2013; Olivié and Peters, 2013; Smith et al. , 2018). Figure 6.15 shows the GSAT response for sustained step reduction in emissions of idealised SLCFs with different lifetimes. The response is relative to a baseline with constant emissions, so effects of emissions before the step reduction is not shown. For SLCFs with lifetimes shorter than a few years, the concentrations quickly reach a new steady state and the response time is primarily governed by the thermal inertia and thus the time scales of the climate system. For compounds with lifetime on the order of 10 years (e.g., methane), there is about a 10-year delay in the response during the first decades, compared to compounds with lifetimes less than one year. However on longer time scales the response is determined solely by the time scales of the climate system itself. For CO2 (dashed line in Figure 6.15) the temporal response is very different due to the long time scale for mixing into the deep ocean and therefore a substantial fraction of atmospheric CO2 is only removed on millenium time scales. This means that for SLCFs including methane, the rate of emissions drives the long-term stabilisation, as opposed to CO2 where the long-term effect is controlled by cumulative emissions (Allen et al., 2018b). Methods to compare rates of SLCF emissions with cumulative CO2 emissions are discussed in Chapter 7 (Section 7.6.1.4).
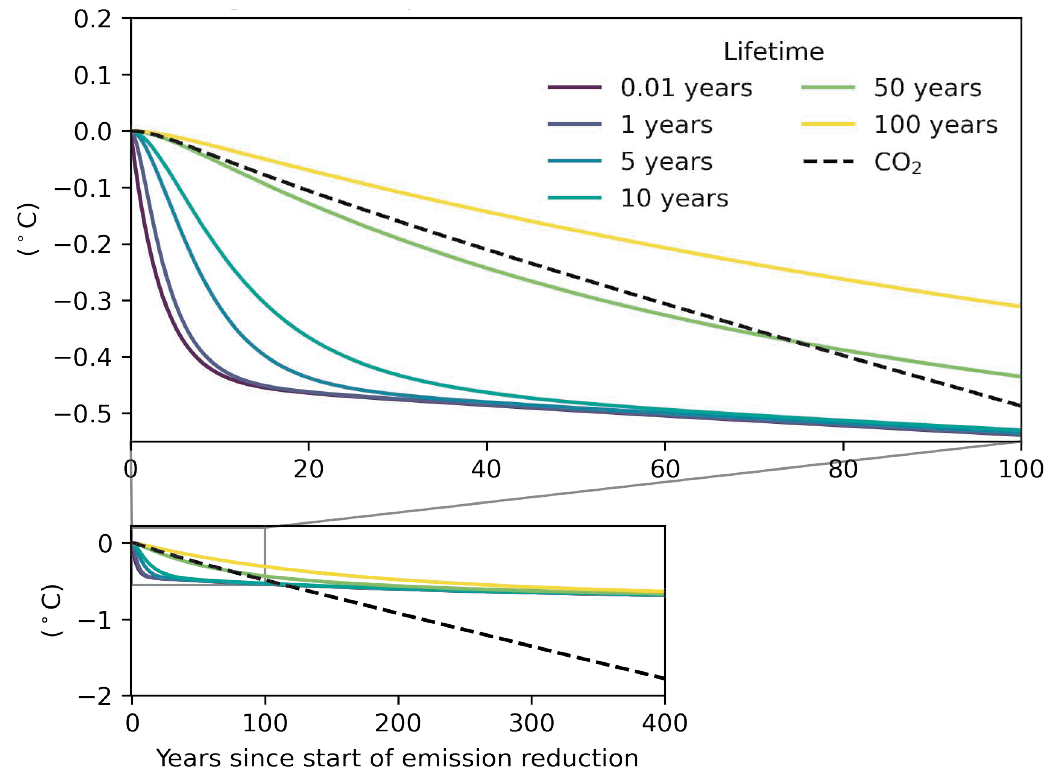 Figure 6.15 | Global mean surface air temperature (GSAT) response to an abrupt reduction in emissions (at time t=0) of idealized climate forcing agents with different lifetimes. All emissions are cut to give a radiative forcing of –1 W m–2 at a steady state (except for CO2). In other words, if the yearly emissions are E0 before the reduction, they will have a fixed lower value Eyear>0= (E0– δE) for all succeeding years. For comparison, the GSAT response to a sustained reduction in CO2 emissions resulting in an RF of –1 W m–2 in year 100 is included (dashed line). The temperature response is calculated using an impulse response function (Cross-Chapter Box 7.1) with a climate feedback parameter of –1.31 W m–2°C–1. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.15 | Global mean surface air temperature (GSAT) response to an abrupt reduction in emissions (at time t=0) of idealized climate forcing agents with different lifetimes. All emissions are cut to give a radiative forcing of –1 W m–2 at a steady state (except for CO2). In other words, if the yearly emissions are E0 before the reduction, they will have a fixed lower value Eyear>0= (E0– δE) for all succeeding years. For comparison, the GSAT response to a sustained reduction in CO2 emissions resulting in an RF of –1 W m–2 in year 100 is included (dashed line). The temperature response is calculated using an impulse response function (Cross-Chapter Box 7.1) with a climate feedback parameter of –1.31 W m–2°C–1. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). As a consequence, in idealized ESM studies that assume an instantaneous removal of all anthropogenic or fossil fuel-related emissions, a rapid change in aerosol levels occurs leading to large increases in GSAT with the rate of warming lasting for several years. Similarly, the thermal inertia causes the pulse emissions (Figure 6.15) of SLCFs to have a significant effect on surface temperature even after 10 years.
In summary, for SLCFs with short lifetime (e.g., months), the response in surface temperature occurs strongly as soon as a sustained change in emissions is implemented and continues to grow for a few years, primarily due to thermal inertia in the climate system (high confidence). Near its maximum, the response slows down but will then take centuries to reach equilibrium (high confidence). For SLCFs with longer lifetimes (e.g., a decade), a delay equivalent to their lifetimes comes in addition to the delay due the thermal inertia (high confidence).
6.6.2 Attribution of Temperature and Air Pollution Changes to Emissions Sectors and Regions
Assessment of the temperature response to source emissions sectors is important for identifying priority mitigation measures and designing efficient mitigation strategies.
Temperature effects of emissions can be quantified for the historical contribution to the present temperature impact (Section 6.4.2), for idealized one-year pulses of emissions or for continued (sustained) emissions at present levels and for changes during a specific time period, for emissions from future scenarios with various hypotheses, giving complementary information to feed mitigation strategies. The AR5 assessed the net global temperature impact of source emissions sectors from a one-year pulse (a single year’s worth) of year 2008 emissions and found that the largest contributors to warming on 50–100-year time scales are the energy, industrial and on-road transportation sectors. Sectors that emit large amounts of methane (agriculture and waste management) and black carbon (residential biofuel) are important contributors to warming over short time horizons up to 20 years. Below, we discuss the effect on ERFs, temperature and air pollution of selected key sectors estimated to have large non-CO2 forcing, including agriculture, residential and commercial, and transport (aviation, shipping, land transportation).
6.6.2.1 Agriculture
According to the SRCCL assessment (Jia et al., 2019), agriculture, forestry and other land use (AFOLU) are a significant net source of GHG emissions (high confidence), with more than half of these emissions attributed to non-CO2 gHGs from agriculture. With respect to SLCFs, agricultural activities are major global sources of methane and NH3 (Section 6.2.1). The agriculture sector exerts strong near-term warming due to large methane emissions that is slightly offset by a small cooling from secondary inorganic aerosols formed notably from the NH3 emissions (Heald and Geddes, 2016; Lund et al., 2020). For present-day emissions, agriculture is the second largest contributor to warming on short time scales but with a small persisting effect on surface temperature (+0.0012°C ± 0.00028°C) after a pulse of current emissions (Figure 6.16, see detailed description in Section 6.6.2.3.4; Lund et al., 2020). Aerosols produced from agricultural emissions, released after nitrogen fertilizer application and from animal husbandry, influence surface air quality and make an important contribution to surface PM2.5 in many densely populated areas ( Figure 6.17; Lelieveld et al., 2015b; Bauer et al., 2016).
6.6.2.2 Residential and Commercial Cooking and Heating
The residential and commercial sector is associated with SLCF emissions of carbonaceous aerosols, CO and NMVOCs, SO2 and NOx, and can be split by fuel type (biofuel or fossil fuel) where residential fossil fuel is also associated with CO2 and methane emissions (Section 6.2.1).
The net effect of residential CO and NMVOC emissions is warming and that of SO2 and NOx is cooling of the atmosphere. However, the sign of the net global radiative effects of carbonaceous aerosols from the residential sector and solid-fuel cookstove emissions (warming or cooling) is not well constrained based on evidence from recent global atmospheric modelling studies. Estimates of direct aerosol–radiation and aerosol–cloud effects fromtheglobal residential sector range from –20 to +60 mW m–2 (Kodros et al., 2015) and –66 to +21 mW m–2 (Butt et al., 2016) and from –20 to +10 mW m–2 (Kodros et al., 2015) and –52 to –16 mW m–2 (Butt et al., 2016), respectively. Uncertainties are due to assumptions about the aerosol emissions masses, size distribution, aerosol optical properties and mixing states (Section 6.3.5.3). Allowing BC to act as an INP in a global model leads to a much larger global forcing estimate from –275 to +154 mW m–2 with a large uncertainty range due to uncertainty in the plausible range of maximum freezing efficiency of BC (Huang et al. , 2018). The residential biofuel sector is a major concern for indoor air quality (Bonjour et al. , 2013). In addition, several atmospheric modelling studies find that this sector is also important for outdoor air quality and even a dominant source of population-weighted outdoor PM2.5 in India and China (Lelieveld et al. 2015b, Silva et al. , 2016; Spracklen et al. , 2018; Reddington et al. , 2019).
The net climate effect of a one-year pulse of current emissions from the residential sector is warming in the near term of +0.0018°C ± 0.00084°C from fossil fuel use and +0.0014°C ± 0.0012°C from biofuel use. Over a 100-year time horizon, this warming is +0.0017°C ± 0.00017°C and +0.0001°C ± 0.000079°C, respectively (Lund et al., 2020). This is due to the effects of BC, methane, CO and NMVOCs, which add to that of CO2, but the uncertainty in the sign of carbonaceous aerosol net effects challenges overall quantitative understanding of this sector and leads to low confidence in this assessment. Residential sector emissions are an important source of indoor and outdoor air pollution in Asia and globally (high confidence).
6.6.2.3 Transportation
6.6.2.3.1 Aviation
Aviation is associated with a range of SLCFs, in particular emissions of NOx and aerosol particles, alongside emissions of water vapour and CO2. The largest SLCF effects are those from the formation of persistent condensation trails (contrails) and NOx emissions. Persistent contrails are ice-crystal clouds, formed around aircraft soot particles (and water vapour from the engine), injected in ambient cold and ice-supersaturated atmosphere, which can spread and form contrail cirrus clouds. The ‘net NOx’ effect arises from the formation of tropospheric ozone, counterbalanced by the destruction of ambient methane and associated cooling effects of reductions in stratospheric water vapour and background ozone. The AR5 assessed the radiative forcing from persistent linear contrails to be +0.01 [+0.005 to +0.03] W m–2 for the year 2011, with medium confidence (Boucher et al., 2013). The combined linear contrail and their subsequent evolution to contrail cirrus radiative forcing from aviation was assessed to be +0.05 [+0.02 to +0.15] W m–2, with low confidence. An additional forcing of +0.003 W m–2 due to emissions of water vapour in the stratosphere by aviation was also reported (Boucher et al., 2013). The aviation sector was also estimated to lead to a net surface warming at 20- and 100-year horizons following a one-year pulse emission. This net temperature response was determined by similar contributions from contrails, contrail cirrus and CO2 over a 20-year time horizon, and dominated by CO2 in a 100-year perspective (Figure 8.34 in AR5, Myhre et al., 2013).
Our assessment is built upon Lee et al. (2021). Their study consists of an updated, comprehensive assessment of aviation climate forcing in terms of RF and ERF based on a large number of studies and the most recent air-traffic and fuel-use datasets available (for 2018), new calculations and the normalization of values from published modelling studies, and combining the resulting best estimates via a Monte-Carlo analysis. Lee et al. (2021) report a net aviation ERF for year-2018 emissions of +0.101 [0.055–0.145] W m–2 with major contributions from contrail cirrus (0.057 W m–2), CO2 (0.034 W m–2) and NOx (0.017 W m–2). Contrails and aviation-induced cirrus yield the largest individual positive ERF followed by CO2 and NOx emissions (Lee et al. 2021). The confidence level in ERF due to contrails and aviation-induced cirrus is assessed to be low in Chapter 7 (Section 7.3.4.2) due to potential missing processes. The formation and emission of sulphate aerosols yield a negative (cooling) term. SLCF forcing terms contribute about eight times more than CO2 to the uncertainty in the aviation net ERF in 2018 (Lee et al., 2021). The largest uncertainty in assessing aviation climate effects is on the interactions of BC and sulphate aerosols on cirrus and mixed-phase clouds, for which no best estimates of the ERFs were provided (Lee et al., 2021).
One of the most significant changes between AR5 and AR6 in terms of aviation SLCFs is the explicit calculation of a contrail cirrus ERF found to be 35% of the corresponding RF (Bickel et al. , 2020), confirming the studies indicating smaller efficacy of linear contrails(Ponater et al., 2005; Rap et al., 2010). The net-NOx term is generally agreed to be a positive RF in the present day, although attribution in a non-linear chemical system is problematic (Grewe et al., 2019), but Skowron et al. (2021) point out that the sign of net NOx term is dependent on background conditions and could be negative under certain future scenarios.
The best estimate ERFs from aviation (Lee et al., 2021) have been used to calculate aviation-specific Absolute Global Temperature change Potential (AGTP) using the method described in Lund et al. (2020) and subsequently compute the effect of a one-year pulse of aviation emissions on global mean surface temperature on a 10- and 100-year time horizon (Section 6.6.2.3.4 and Figure 6.16). The effect of contrail-cirrus is most important for the estimated net-GSAT response after the first decade, followed by similar warming contributions from NOx and CO2 emissions. At a 20-year time horizon, the net contribution from aviation to GSAT has switched from a positive to a small negative effect (see Supplementary Material 6.SM.4). This is due to the combination of rapidly decaying contrail-cirrus warming and the complex time variation of the net temperature response to NOx emissions, which changes sign between 10 and 20 years due to the balance between the positive short-lived ozone forcing and negative forcing from changes in methane and methane-induced changes in ozone and stratospheric water vapour. The net GSAT response to aviation emissions has previously been estimated to be positive on a 20-year time horizon (AR5, Chapter 8; Lund et al., 2017). This difference in net GSAT after 20 years in AR5 compared to AR6, results primarily from a shorter time scale of the climate response in the underlying AGTP calculations in Lund et al. (2020), which means the initial, strong impacts of the most short-lived SLCFs, including the warming by contrail-cirrus decay faster, in turn giving the net NOx effect a relatively higher importance after 20 years. On longer time horizons, the net GSAT response switches back to positive, as CO2 becomes the dominating warming contribution.
In summary, the net aviation ERF is assessed to be +0.1 W m–2 (±0.045) for the year 2018 (low confidence). This confidence level is largely a result of the fact that the SLCF-related terms, which account for more than half (66%) of the net aviation ERF, are the most uncertain terms. The climate response to SLCF-related aviation terms exhibits substantial spatio-temporal heterogeneity in characteristics (high confidence). Overall, cirrus and contrail cirrus warming, as well as NOx-induced ozone increase, induce strong but short-lived warming contributions to the GSAT response 10 years after a one-year pulse of present-day aviation emissions (medium confidence), while CO2 both gives a warming effect in the near term and dominates the long-term warming impact (high confidence).
6.6.2.3.2 Shipping
Quantifying the effects of shipping on climate is particularly challenging because (i) the sulphate cooling impact is dominated by aerosol–cloud interactions and (ii) ship emissions contain NOx, SOx and BC, which lead to mixed particles. Previous estimates of the sulphate radiative effects from present-day shipping span the range –47 to –8 mW m–2 (direct radiative effect) and –600 to –38 mW m–2 (indirect radiative effects) (Lauer et al. , 2007; Balkanski et al. , 2010; Eyring et al. , 2010; Lund et al. , 2012). Partanen et al. (2013) reported a global mean ERF for year-2010 shipping aerosol emissions of –390 mW m–2. The temperature change has been shown to be highly sensitive to the choice of aerosol–cloud parametrization (Lund et al., 2012). One year of global present-day shipping emissions, not considering the impact of recent low sulphur fuel regulation (IMO, 2016), are estimated to cause net cooling in the near term (–0.0024°C ± 0.0025°C) and slight warming (+0.00033°C ± 0.00015°C) on a 100-year horizon (Lund et al., 2020).
Shipping is also of importance for air pollution in coastal areas along the major trade routes, especially in Europe and Asia (Corbett et al. , 2007; H. Liu et al. , 2016, Figure 6.17; Jonson et al. , 2020). Jonson et al. (2020) estimated that shipping is responsible for 10% or more of the controllable PM2.5 concentrations and depositions of oxidised nitrogen and sulphur for many coastal countries. Widespread introduction of low-sulphur fuels in shipping from 2020 (IMO, 2016) will lead to improved air quality and reduction in premature mortality and morbidity (Sofiev et al., 2018).
In summary, a year’s worth of present-day global shipping emissions (i.e., without the implementation of the 2020 clean fuel standards) cause a net global cooling (–0.0024 ± 0.0025°C) on 10–20 year time horizons (high confidence) but its magnitude is of low confidence.
6.6.2.3.3 Land transportation
The on-road and off-road transportation sectors have a net warming impact on climate over all time scales (Berntsen and Fuglestvedt, 2008; Fuglestvedt et al., 2008; Unger et al., 2010; Lund et al., 2020). A one-year pulse of present-day emissions has a small net global temperature effect on short time scales (+0.0011°C ± 0.0045°C), predominantly driven by CO2 and BC warming offset by NOx-induced cooling through methane lifetime reductions (Lund et al., 2020).
The vehicle tailpipe emissions profiles of diesel and gasoline are distinctly different. Diesel air pollutant emissions are dominated by BC and NOx whereas gasoline air pollutant emissions are dominated by CO and NMVOCs, especially when distribution and upstream losses are considered. Thus, the net radiative effect of the on-road vehicle fleets depends upon the share of different fuels used, in particular gasoline and diesel (Lund et al., 2014; Huang et al., 2020). The net SLCF for year-2010 emissions from the global diesel vehicle fleet have been estimated to be +28 mW m–2 (Lund et al., 2014). Huang et al. (2020) estimated net global radiative effects of SLCFs (including aerosols, ozone, and methane) from the gasoline and diesel vehicle fleets in the year 2015 to be +13.6 and +9.4 mW m–2, respectively, with similar fractional contributions of SLCFs to the total global climate impact including CO2 on the 20‐year time scale (14–15%).
There is consensus that on‐road transportation sector emissions, including gasoline and diesel, are important anthropogenic contributors to elevated surface ozone and PM2.5 concentrations (Chambliss et al. , 2014; Lelieveld et al., 2015b ; Silva et al. , 2016; Anenberg et al. , 2019). At a global scale, land transportation has been estimated to be the dominant contributor to surface ozone concentrations in populated areas (Silva et al., 2016) and ozone-induced vegetation damages (Section 6.4.4; Unger et al., 2020). Furthermore, it is now well established that real-world diesel NOx emissions rates are substantially higher, the so-called ‘excess NOx’, in all regional markets than in laboratory tests, worsening air quality (Anenberg et al., 2017; Jonson et al., 2017; Chossière et al., 2018) and contributing to slightly larger warming on the scale of years and smaller warming at the decadal scale (Tanaka et al., 2018). Excess NOx emissions from key global diesel markets are estimated at 4.6 Tg yr–1 in 2015, with annual mean ozone and PM2.5 increases of 1 ppb and 1µg m–3across large regions of Europe, India and China (Anenberg et al., 2017).
In summary, the present-day global land-based transport pulse emissions cause a net global warming on all time scales (high confidence) and are detrimental to air quality (high confidence).
6.6.2.4 GSAT Response to Emissions Pulse of Current Emissions
Figure 6.16 presents the GSAT response to an idealized pulse of year-2014 emissions of individual SLCF and LLGHG. The GSAT response is calculated for 11 sectors and 10 regions accounting for best available knowledge and geographical dependence of the forcing efficacy of different SLCFs (Lund et al., 2020). Two time horizons are shown (of 10 and 100 years) to represent near- and long-term effects (and a 20-year horizon is presented in Supplementary Material Figure 6.SM.3). Other time-horizon choices may affect the relative importance, and even sign in the case of the NOx effect, of the temperature response from some of the SLCFs, or be more relevant for certain applications. GSAT response is calculated using the concept of AGTP (Section 7.6.2.2). Further details of the AGTP emulator applied in Figure 6.16 are provided in Lund et al. (2020) and Supplementary Material 6.SM.4 (Section 7.6.1.2, Cross-Chapter Box 7.1 and Supplementary Material 7.SM.7.2). As discussed by Lund et al. (2020), the AGTP framework is primarily designed to study the relative importance of individual emissions and sources, but the absolute magnitude of temperature responses should be interpreted with care due to the linearity of the AGTP, which does not necessarily capture all the non-linear effects of SLCFs emissions on temperature.
Differences in the mix of emissions result in net effects on GSAT that vary substantially, in both magnitude and sign, between sectors and regions. SLCFs contribute substantially to the GSAT effects of sectors on short time horizons (10–20 years) but CO2 dominates on longer time horizons (Figure 6.16). As the effect of the SLCFs decays rapidly over the first few decades after emission, the net long-term temperature effect is predominantly determined by CO2. N2O adds a small contribution to the long-term effect of agriculture. CO2 emissions cause an important contribution to near-term warming that is not always fully acknowledged in discussions of LLGHGs and SLCFs (Lund et al., 2020).
The global sectoral ranking for near- and long-term global temperature effects is similar to the AR5 assessment despite regional reductions in aerosol precursor emissions between 2008 and 2014. This report has applied updated climate policy metrics for SLCFs and treatment of aerosol–cloud interactions for SO2, BC and OC (Lund et al., 2020). By far the largest 10-year GSAT effects are from the energy production (fossil fuel mining and distribution), agriculture and waste management sectors (high confidence). Methane is the dominant contributor in the energy production, agriculture and waste management sectors. On the 10-year time horizon, other net warming sectors are residential fossil fuel and energy combustion (dominated by CO2) and aviation and residential biofuel (dominated by SLCFs and cloud) (medium confidence). The total residential and commercial sector, including biofuel and fossil fuels, is the fourth largest contributor to warming globally on short time horizons of 10–20 years. The energy combustion sector has considerable cooling from high emissions of SO2 that result in a relatively small net GSAT temperature effect on short time horizons, despite the high CO2 emissions from this activity. On the 10-year time horizon, global emissions from industry and shipping cause a net cooling effect despite a considerable warming from CO2 emissions. On the 100-year time horizon, the net effects of agriculture and waste management are small, while energy combustion is the largest individual contributor to warming due to its high CO2 emissions. The second largest driver of long-term temperature change is industry, demonstrating the importance of non-CO2 emissions for shaping relative weight over different time frames. Transport contributes a small net warming on the 10-year time horizon that increases by a factor of three on the 100-year time horizon. In contrast, the aviation sector contribution to warming shrinks by about a factor of three between the 10- and 100-year time horizons. Results for the 20-year time horizon are provided in the Supplementary Material 6.SM.4. Compared to the 10-year time horizon, there are some changes in ranking, especially of sectors and regions with a strong SO2 contribution, which decays substantially between 10 and 20 years. Aviation is the sector with the most distinct difference between 10- and 20-year time horizons, such that the net GSAT effect after 20 years becomes small but negative. This is due to a switch in sign for the NOx AGTP for this sector and the stronger effect of short-lived ozone response over these two short-term horizons in the case of aviation compared with other sectors.
In terms of source regions, the largest contributions to net short-term warming are caused by emissions in Eastern Asia, Latin America and North America, followed by Africa, Eastern Europe, West-Central Asia and South East Asia (medium confidence). However, the relative contributions from individual species vary. In Eastern Asia, North America, Europe and Southern Asia, the effect of current emissions of cooling and warming SLCFs approximately balance in the near term and these regions cause comparable net warming effects on 10- and 100-year time horizons (Figure 6.16). In Latin America, Africa, and South East Asia and Developing Pacific, methane and BC emissions are currently high while emissions of CO2 and cooling aerosols are low compared to other regions, resulting in a net warming effect after 10 years that is substantially higher than that of CO2 alone.
Overall, the global sectors that contribute the largest warming on short time scales are the methane-dominated sources, that is energy production (fossil fuel mining and distribution), and agriculture and waste management (high confidence). On short time scales, other net warming sectors are residential fossil fuel and energy combustion (dominated by CO2), and aviation and residential biofuel (dominated by SLCFs) (medium confidence). On short time scales, global emissions from industry and shipping cause a net cooling effect despite a considerable warming from CO2 emissions (high confidence). On longer time horizons, the sectors that contribute the largest warming are energy combustion and industry due to the large CO2 emissions (high confidence).
6.6.2.5 Source attribution of regional air pollution
The attribution of present-day surface PM2.5 and ozone concentrations to sectors and regions (Figure 6.17) is based on 2014 CMIP6 emissions used in the TM5-FASST model (Van Dingenen et al., 2018) that has been widely applied to analyse air quality in regional and global scenarios (e.g., Van Dingenen et al. , 2009; Rao et al. , 2016, 2017; Vandyck et al. , 2018; Harmsen et al. , 2020b). Regions with the largest year-2014 population-weighted annual average surface PM2.5 concentrations are Southern Asia, Eastern Asia and the Middle East. The dominant anthropogenic source of ambient PM2.5 in Southern Asia are the residential and commercial sectors (biomass and coal fuel-based cooking and heating) with secondary contributions from energy and industry. In Eastern Asia, the main anthropogenic sources of ambient PM2.5 are energy, industry and residential sources. Natural sources, predominantly dust, are the most important PM2.5 source in the Middle East, Africa and Eurasia, contributing about 40–70% of ambient annual average concentrations (Figure 6.17). Agriculture is an important contributor to ambient PM2.5 in Europe and North America, while open biomass burning is a major contributor in South East Asia and Developing Pacific, North America as well as Latin America. These results are consistent with several global and regional studies, where contribution of emissions sources to ambient PM2.5 or premature mortality was estimated at different scales (e.g., Guttikunda et al. , 2014; Lelieveld et al. 2015b ,; Amann et al. , 2017; Qiao et al. , 2018; Venkataraman et al. , 2018; Wu et al. , 2018).
Natural sources contribute more than 50% to surface ozone in all regions except Southern Asia and South East Asia. Southern Asia, Eastern Asia and the Middle East experience the highest surface ozone levels of all regions. For ozone, the anthropogenic sectoral attribution is more uniform across regions than for PM2.5, except for Southern and South East Asia, where land transportation plays a larger role, and Eastern Asia, where the most significant contribution is from energy and industry. Land transportation and energy are the most important contributors to ozone across many of the regions, with smaller contributions from agriculture, biomass burning, waste management and industry. Open biomass burning is not a major contributor to surface ozone, except for in Africa, Latin America and South East Asia where its contribution is estimated at about 5–10% of anthropogenic sources. The relative importance of natural and anthropogenic emissions sources on surface ozone has been assessed in several studies (Uherek et al., 2010; Zare et al., 2014; Mertens et al., 2020; Unger et al., 2020) and the results are comparable with the estimates of the TM5-FASST used here.
Residential and commercial cooking and heating are among the most important anthropogenic sources of ambient PM2.5, except in the Middle East and Asia-Pacific Developed (high confidence) and agriculture is the dominant source in Europe and North America (medium confidence). Energy and industry are important PM2.5 contributors in most regions, except Africa (high confidence). Energy and land transportation are the major anthropogenic sources of ozone across many world regions (medium to high confidence).
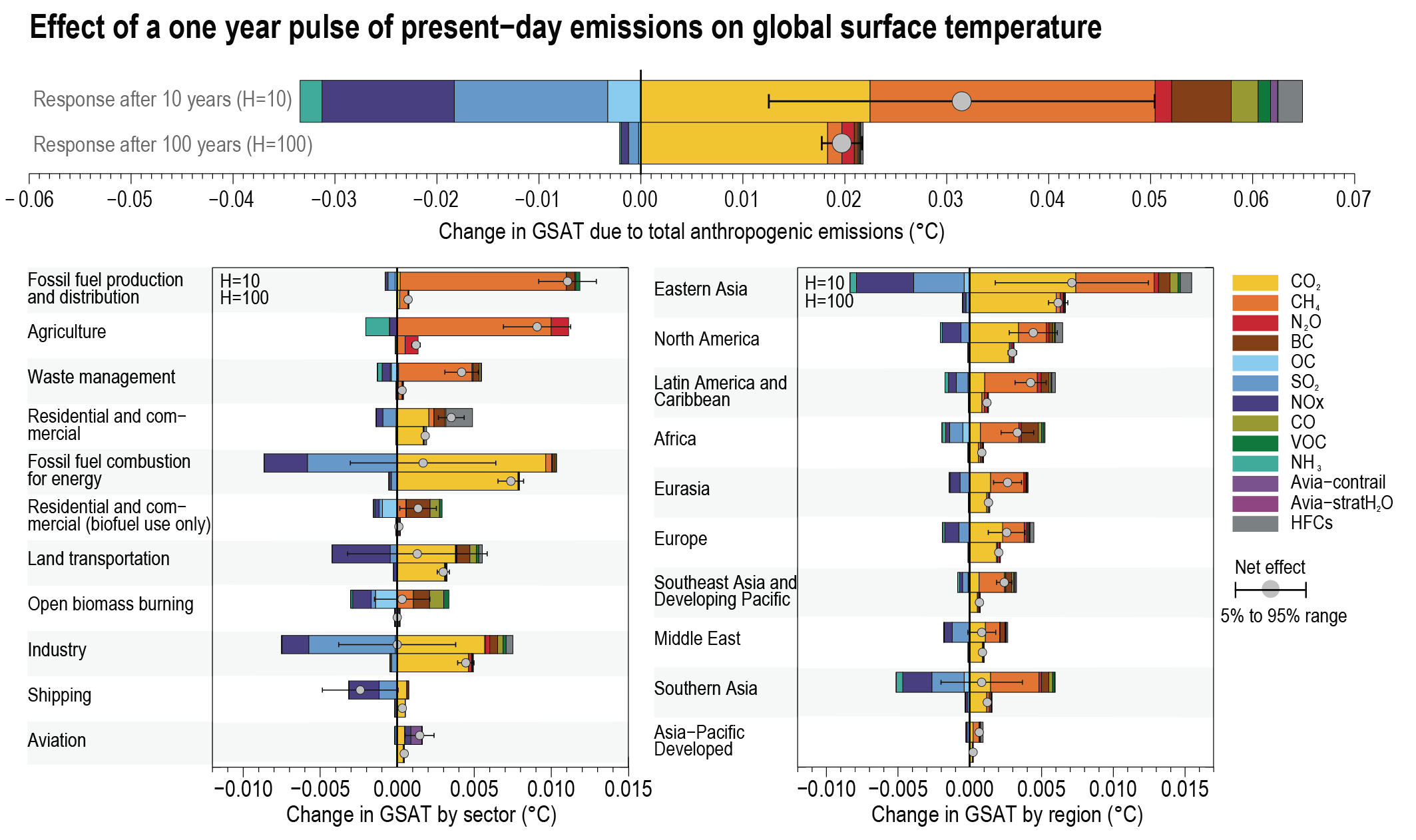 Figure 6.16 | Global mean temperature response 10 and 100 years following one year of present-day (year 2014) emissions. The temperature response is broken down by individual species and shown for total anthropogenic emissions (top), sectoral emissions (left) and regional emissions (right). Sectors and regions are sorted by (high-to-low) net temperature effect on the 10-year time scale. Error bars in the top panel show uncertainty (5–95% interval) in net temperature effect due to uncertainty in radiative forcing only (calculated using a Monte Carlo approach and best estimate uncertainties from the literature – see Lund et al. (2020) for details). CO2 emissions are excluded from open biomass burning and residential biofuel use due to their unavailability in the Community Emissions Data System (CEDS) and uncertainties around non-sustainable emission fraction. Emissions for 2014 originate from the CEDS (Hoesly et al., 2018), except for HFCs which are from Purohit et al. (2020), open biomass burning from van Marle et al. (2017), and aviation H2O which is from Lee et al. (2021). The split of fossil fuel production and distribution and combustion for energy and residential and commercial fuel use into fossil fuel and biofuel components is obtained from the GAINS model (ECLIPSE version 6b dataset). Open biomass burning emissions are not included for the regions. Emissions are aggregated into fossil fuel production and distribution (coal mining, oil and gas production, upstream gas flaring and gas distribution networks), agriculture (livestock and crop production), fossil fuel combustion for energy (power plants), industry (combustion and production processes, solvent-use losses from production and end use), residential and commercial (fossil fuel use for cooking and heating as well is HFCs leakage from A/C and refrigeration), waste management (solid waste, including landfills and open trash burning, residential and industrial waste water), transport (road and off-road vehicles, and HFC leakage from A/C and refrigeration equipment), residential and commercial (biofuels use for cooking and heating), open biomass burning (forest, grassland, savanna fires and agricultural waste burning), shipping (including international shipping), and aviation (including international aviation). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.16 | Global mean temperature response 10 and 100 years following one year of present-day (year 2014) emissions. The temperature response is broken down by individual species and shown for total anthropogenic emissions (top), sectoral emissions (left) and regional emissions (right). Sectors and regions are sorted by (high-to-low) net temperature effect on the 10-year time scale. Error bars in the top panel show uncertainty (5–95% interval) in net temperature effect due to uncertainty in radiative forcing only (calculated using a Monte Carlo approach and best estimate uncertainties from the literature – see Lund et al. (2020) for details). CO2 emissions are excluded from open biomass burning and residential biofuel use due to their unavailability in the Community Emissions Data System (CEDS) and uncertainties around non-sustainable emission fraction. Emissions for 2014 originate from the CEDS (Hoesly et al., 2018), except for HFCs which are from Purohit et al. (2020), open biomass burning from van Marle et al. (2017), and aviation H2O which is from Lee et al. (2021). The split of fossil fuel production and distribution and combustion for energy and residential and commercial fuel use into fossil fuel and biofuel components is obtained from the GAINS model (ECLIPSE version 6b dataset). Open biomass burning emissions are not included for the regions. Emissions are aggregated into fossil fuel production and distribution (coal mining, oil and gas production, upstream gas flaring and gas distribution networks), agriculture (livestock and crop production), fossil fuel combustion for energy (power plants), industry (combustion and production processes, solvent-use losses from production and end use), residential and commercial (fossil fuel use for cooking and heating as well is HFCs leakage from A/C and refrigeration), waste management (solid waste, including landfills and open trash burning, residential and industrial waste water), transport (road and off-road vehicles, and HFC leakage from A/C and refrigeration equipment), residential and commercial (biofuels use for cooking and heating), open biomass burning (forest, grassland, savanna fires and agricultural waste burning), shipping (including international shipping), and aviation (including international aviation). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). 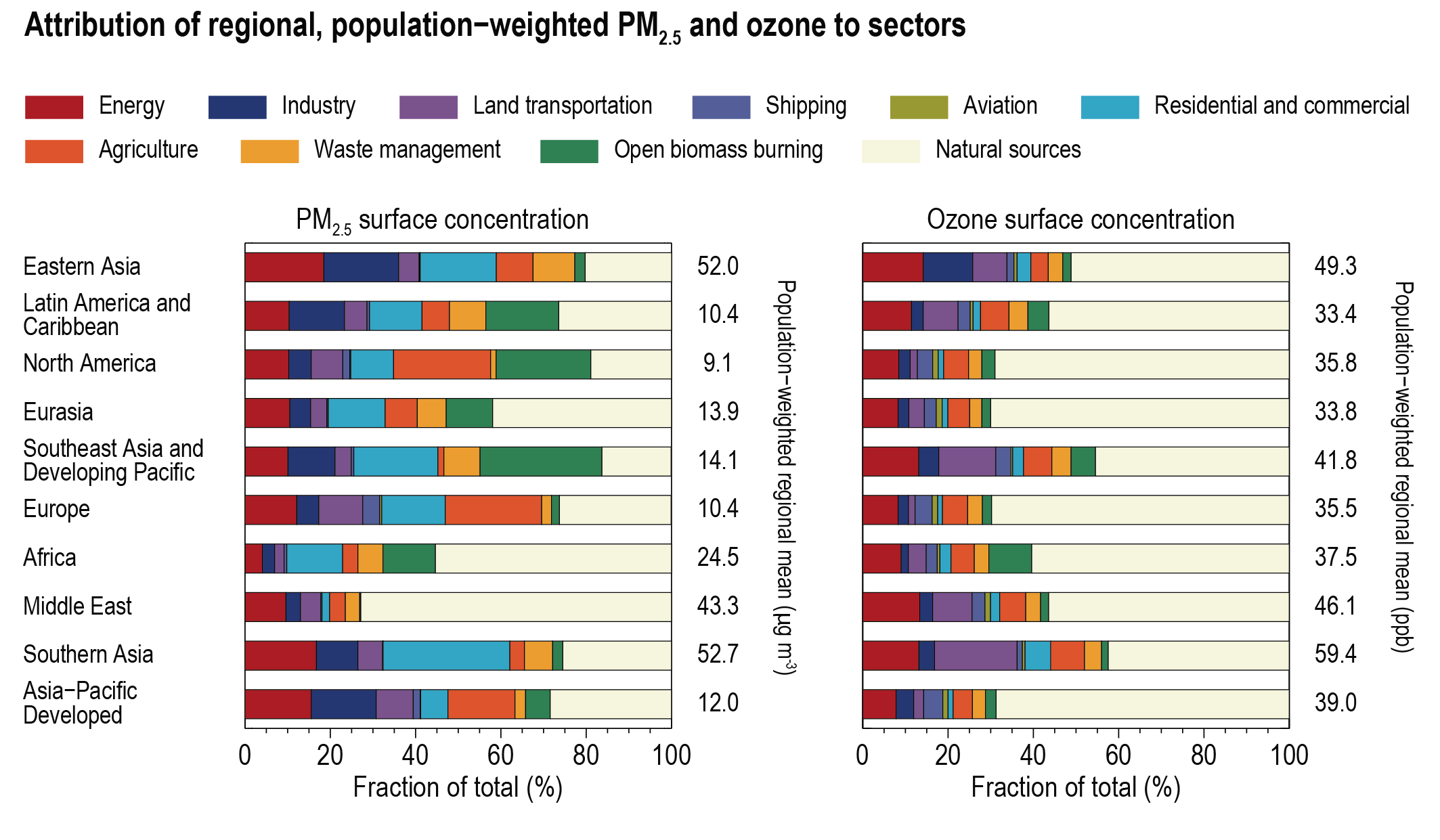 Figure 6.17 | Emissions source-sector attribution of regional population-weighted mean concentrations of PM2.5and ozone for present-day emissions (year 2014). Regional concentrations and source apportionment are calculated with the TM5-FASST model (Van Dingenen et al., 2018) for the 2014 emissions data from the Community Emissions Data System (CEDS) (Hoesly et al., 2018) and van Marle et al. (2017) for open-biomass burning. Dust and sea salt contributions to PM2.5 are monthly mean climatological averages over 2010–2018 from CAMS global reanalysis (EAC4) (Inness et al., 2019), generated using Copernicus Climate Change Service information (January 2020). Anthropogenic sectors are similar to those in Figures 6.2 and 6.16, except the grouping of fossil fuel production, distribution and combustion for energy under ‘Energy’ and the grouping of use of fossil fuel and biofuel use for cooking and heating under ‘Residential and Commercial’. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.17 | Emissions source-sector attribution of regional population-weighted mean concentrations of PM2.5and ozone for present-day emissions (year 2014). Regional concentrations and source apportionment are calculated with the TM5-FASST model (Van Dingenen et al., 2018) for the 2014 emissions data from the Community Emissions Data System (CEDS) (Hoesly et al., 2018) and van Marle et al. (2017) for open-biomass burning. Dust and sea salt contributions to PM2.5 are monthly mean climatological averages over 2010–2018 from CAMS global reanalysis (EAC4) (Inness et al., 2019), generated using Copernicus Climate Change Service information (January 2020). Anthropogenic sectors are similar to those in Figures 6.2 and 6.16, except the grouping of fossil fuel production, distribution and combustion for energy under ‘Energy’ and the grouping of use of fossil fuel and biofuel use for cooking and heating under ‘Residential and Commercial’. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). 6.6.3 Past and Current SLCF Reduction Policies and Future Mitigation Opportunities
Several SLCF-emissions reduction strategies have been explored in the literature or are already pursued as part of environmental and development policies, including air quality, waste management, energy poverty and climate change. The effects of various policies and strategies have been addressed in a limited number of modelling studies with different objectives that range from assessment of specific policies and their regional effects (UNEP and WMO, 2011; Shindell et al., 2012, 2017b; AMAP, 2015a, b; Haines et al., 2017; UNEP and CCAC, 2018; UNEP, 2019; Harmsen et al. , 2020a) to large-scale global-scenario studies with varying levels of SLCF control (e.g., Sand et al., 2016; Rogelj et al., 2018b; Shindell and Smith, 2019). They could be grouped into:
- Projections of future SLCF emissions compatible with the climate change mitigation trajectories investigated in the climate model intercomparison projects (respective RCP or SSP scenarios): in such scenarios, when climate change mitigation is considered, it is associated with a strong decrease of CO2 emissions, largely relying on fossil fuel-use reduction, along with proportional reductions in the co-emitted SLCFs from combustion and methane from the production and distribution of fossil fuels. Depending on the carbon price and climate change mitigation target, further reduction of methane from waste and agriculture will also be part of such scenarios. The limitations of RCP scenarios (where continuous strengthening of air-quality legislation was assumed resulting in lack of futures where global and regional air quality deteriorates) for the analysis of air quality and potential for mitigation of SLCFs have been discussed in literature (e.g., Amann et al., 2013; von Schneidemesser et al., 2015). SSP scenarios consider various levels of air pollution control, in accordance with their socio-economic narrative, and thus cover a wider span of SLCF trajectories (Section 6.7). The economic cost of the implementation of these scenarios and their co-benefits on air quality and SDGs are assessed in the AR6 WGIII Report (Chapter 3).
- Projections of SLCF emissions assuming strong reduction of all air pollutants in the absence of climate change mitigation (e.g., the SSP3-lowSLCF scenario): the latter is an idealized simulation of a very ambitious air-quality policy where the maximum technical potential of existing end-of-pipe technologies is explored in the SSP3-7.0 scenario. Methane reduction can also be part of such sensitivity analysis, although methane reductions have not historically been motivated by air pollution concerns.
- Projection of emissions targeting air quality or other development priorities: anthropogenic emissions’ source-structure and the level of exposure to pollution and subsequent effects varies significantly from one region to another. Therefore, air-quality policies, regional climate impact concerns and development priorities, as well as the consequent level of mitigation of particular SLCF species, will differ regionally and source-wise with respect to the emissions sources, influence of inter-continental transport of pollution, and spatial physical heterogeneities (Lund et al. , 2014; AMAP, 2015a; Sand et al. , 2016; Turnock et al. , 2016; Sofiev et al. , 2018; WMO, 2018). This is also the case at a finer local and regional scale where priorities and scope for SLCF mitigation will differ (e.g., Amann et al., 2017; UNEP, 2019).
- Projections exploring mitigation potential for a particular source or SLCF: These studies focus on the assessment of SLCF reduction potential that can be realised with either existing and proven technologies or extend the scope to include transformational changes needed to achieve further reduction (e.g., UNEP and WMO, 2011; Stohl et al., 2015; Velders et al., 2015; Purohit and Höglund-Isaksson, 2017; Gómez-Sanabria et al., 2018; UNEP, 2019; Höglund-Isaksson et al., 2020; Purohit et al., 2020) – several of these studies are subsequently used for parametrization of the models used to develop emissions scenarios (e.g., IAM models used in the IPCC process).
In the following subsections, we assess the SLCF mitigation and its effects as identified in regional and global studies evaluating past and current air quality and other SLCF regulations (Sections 6.6.3.1, 6.6.3.2 and 6.6.3.3). Development policies, independent from the CMIP6 assessment framework, including peer-reviewed studies and initiatives like the United Nations Environment Programme (UNEP), analysing win-win climate, air-quality and SDG-motivated strategies are discussed in Section 6.6.3.4. Note that sensitivity studies where impacts of complete removal of particular species are analysed (e.g., Samset et al., 2018) are used sparingly in this assessment. While such analysis can be useful for assessing the effect of a zero-emissions commitment (Chapter 4.7.1.1), they do not correspond to a realistic SLCF-mitigation strategy with plausible pace of implementation and removal of co-emitted species (Shindell and Smith, 2019). Discussion of climate and air-quality implications of SLCF reductions in SSP scenarios is provided in Section 6.7.
6.6.3.1 Climate Response to Past AQ Policies
Air-quality policies emerged several decades ago focusing on emissions mitigation, first driven by local- then by regional-scale air-quality and ecosystem-damage concerns, that is, health impacts, acidification and eutrophication. They have made it possible to reduce or limit pollution exposure in many megacities or highly populated regions, for example, in Los Angeles, Mexico City and Houston in North America (Parrish et al., 2011), Santiago in Chile (Gallardo et al., 2018), São Paulo in Brazil (Andrade et al., 2017), Europe (Reis et al., 2012; Crippa et al., 2016; Serrano et al., 2019), and over Eastern Asia during the last decade (Silver et al., 2018; Zheng et al., 2018b). However, very few studies have quantified the impact of these policies on climate. The AR5 concluded that air-quality control will have consequences on climate including strong regional variability, however, no estimates of impacts of specific air-quality policy were available. Since AR5, few studies have provided estimates of climate-relevant indicators affected by significant air pollutant burden changes due to air-quality policy in selected regions. Turnock et al. (2016) estimated that the strong decrease in NOx, SO2 and PM2.5 emissions in Europe, induced by air-quality policies resulting in implementation of abatement measures since the 1970s, have caused a surface warming of +0.45°C ± 0.11°C and increase of precipitation +13 ± 0.8 mm yr–1 over Europe, compared to the scenario without such policies. While the temperature increase is likely overestimated since the impact of the increase in ammonium nitrate was not considered in this study, the simulated European all-sky TOA radiative effect of the European air pollutant mitigation over the period 1970–2009 is 2.5 times the change in global mean CO2 radiative forcing over the same period (Myhre et al., 2013). Other studies found that the recent measures to reduce pollution over China have induced a decrease of aerosols and increase of ozone over east China (K. Li et al., 2019, 2020), resulting in an overall warming effect mainly due to the dominant effect of sulphate reductions in the period 2012–2017 (Dang and Liao, 2019).
6.6.3.2 Recently Decided SLCF-relevant Global Legislation
International shipping emissions regulation: from January 2020, a new global standard, proposed by the International Maritime Organisation, limits the sulphur content in marine fuels to 0.5% against the previous 3.5% (IMO, 2016). This legislation is considered in the SSP5 and SSP2-4.5 and with a delay of few years in SSP3-lowSLCF, SSP1-1.9, and SSP1-2.6, and in other SSP-emissions scenarios achieved by the mid-21st century. This global measure aims to reduce the formation of sulphate (and consequently PM2.5) and largely reduce the health exposure to PM2.5, especially over India, east China and coastal areas of Africa, and the Middle East (Sofiev et al., 2018). Sofiev et al. (2018) used a high spatial-and-temporal resolution chemistry climate model and estimated a net total ERF of +71 mW m–2 Associated with this measure and due to lower direct aerosol cooling (+3.9 mW m–2) and lower cloud albedo (+67 mW m–2). This value, which correponds to an 80% decrease of the cooling effect of shipping induced by about 8 Tg of SO2 of avoided emissions, is consistent with older estimates which considered similar reduction of emitted sulphur. However, there is considerable uncertainty in the indirect forcing since small changes in aerosols, acting as CCNs in a clean environment, can have disproportionally large effects on the radiative balance. Since sulphate is by far the largest component of the radiative forcing (Fuglestvedt et al., 2008) and of surface temperature effect (Figure 6.16) due to ship emissions over a short time scale, limiting the co-emitted SLCFs can not offset the warming by sulphur reductions. The reduction of sulphur emissions from shipping is assessed to lead to a slight warming mainly due to aerosol–cloud interactions (medium evidence, medium agreement).
The Kigali Amendment (UNEP, 2016): with the adoption of the Kigali Amendment to the Montreal Protocol (UN, 1989) in 2016, parties agreed to the phase-down of HFCs, substances that are not ozone depleting but are climate-forcing agents (Papanastasiou et al., 2018). Baseline scenarios, in the absence of controls or only pre-Kigali national legislation, projected increased use and emissions of HFCs. All recent baseline projections are significantly higher than those used in the Representative Concentration Pathways (RCP) scenarios (Figure 6.18; Meinshausen et al., 2011). There is low confidence that the high baseline (assuming absence of controls, lack of technical progress and high growth) as developed by Velders et al. (2009), resulting in additional warming of about 0.5°C by 2100 (Xu et al., 2013; WMO, 2018), is plausible. Evolution of HFC emissions along the baselines consistent with Velders et al. (2009) and Velders et al. (2015) would result in a global average warming, due to HFCs, relative to 2000, of about 0.1°C–0.12°C by 2050 and 0.35°C–0.5°C and 0.28°C–0.44°C by 2100, respectively (Xu et al., 2013). The baseline implementation considered in SSP5-8.5 (Section 6.7.1.1) is comparable to the lower bound of projections by Velders et al. (2015; Figure 6.18) and several other studies (Gschrey et al., 2011; Purohit and Höglund-Isaksson, 2017; EPA, 2019; Purohit et al., 2020) and result in additional warming of 0.15°C–0.3°C by 2100 (medium confidence) (Figure 6.22).
Efficient implementation of the Kigali Amendment and national and regional regulations has been projected to reduce global average warming in 2050 by 0.05°C–0.07°C (Klimont et al., 2017b; WMO, 2018) and by 0.2°C–0.4°C in 2100 compared with the baseline (see Figure 2.20 of WMO, 2018). Analysis of SSP scenarios based on an emulator (Section 6.7.3) shows a comparable mitigation potential of about 0.02°C–0.07°C in 2050 and about 0.1°C–0.3°C in 2100 (Figure 6.22, SSP5-8.5 versus SSP1-2.6). Furthermore, the energy efficiency improvements of cooling equipment alongside the transition to low-global-warming potential alternative refrigerants for refrigeration and air-conditioning equipment could potentially increase the climate benefits from the HFC phasedown under the Kigali Amendment (Shah et al. ,2015; Höglund-Isaksson et al. , 2017; Purohit and Höglund-Isaksson, 2017; WMO, 2018). Purohit et al. (2020) estimated that depending on the expected rate of technological development, improving the energy efficiency of stationary cooling technologies and compliance with the Kigali Amendment could bring future global electricity savings of more than 20% of the world’s expected electricity consumption beyond 2050 or cumulative reduction of about 75–275 Gt CO2- eq over the period 2018–2100 (medium confidence). This could potentially double the climate benefits of the HFC phase-down of the Kigali Amendment as well as result in small air-quality improvements due to reduced air pollutant emissions from the power sector (i.e., 8–16% reduction of PM2.5, SO2 and NOx; Purohit et al., 2020).
6.6.3.3 Assessment of SLCF Mitigation Strategies and Opportunities
There is a consensus in the literature that mitigation of SLCF emissions plays a central role in simultaneous mitigation of climate change, air quality and other development goals including SDG targets (UNEP and WMO, 2011; Shindell et al. , 2012, 2017b; Rogelj et al. , 2014b, 2018b; AMAP, 2015a; Haines et al. , 2017; Klimont et al. , 2017b; McCollum et al. , 2018; Rafaj et al. , 2018; UNEP and CCAC, 2018; UNEP, 2019). There is less agreement in the literature with respect to the actual mitigation potential (or its potential rate of implementation), necessary policies to trigger successful implementation, and resulting climate impacts. Most studies agree that climate policies, especially those aiming to keep warming below 1.5°C or 2°C, trigger large SLCF mitigation co-benefits, (e.g., Rogelj et al., 2014b, 2018b), however, discussion of practical implementation of respective policies and SDGs has only started (Haines et al., 2017). Note that mitigation scenarios outside of the SSP framework are assessed here while those within the SSPs are assessed in Section 6.7.3.
Focusing on air quality, specifically addressing aerosols, by introducing the best available technology reducing PM2.5, SO2 and NOx in most Asian countries within the 2030–2050 time frame (a strategy that has indeed shown reduction in PM2.5 exposure in China) comes, in many regions, short of national regulatory PM2.5 concentration standards (often set at 35 µg m–3 for annual mean; UNEP, 2019). Similarly, global studies (Rafaj et al., 2018; Amann et al., 2020) show that strengthening current air-quality policies, that address primarily aerosols and their precursors, will not enable the achievement of WHO air quality guidelines (annual average concentration of PM2.54 below 10 µg m–3) in many regions.
A multi-model study (four ESMs and six CTMs) found a consistent response to the removal of SO2 emissions that resulted in a global mean surface temperature increase of 0.69°C (0.4°C–0.84°C). However, results are mixed for a global BC-focused deep SLCF reduction without SO2 and methane mitigation which remain as in the baseline (see ECLIPSE in Figure 6.18). BC contributed about –0.022°C temperature reduction for the decade 2041–2050 based on the assumption that mitigation of the non-methane species contributed only about 10% of the global temperature reduction for the strategy where methane mitigation was also included (–0.22°C ± 0.07°C; Stohl et al., 2015). These results are consistent with studies analysing similar strategies using emulators (e.g., Smith and Mizrahi, 2013; Rogelj et al., 2014b). Stohl et al. (2015) also analysed the impact of BC-focused mitigation on air quality, estimating large-scale regional reduction in PM2.5 mean concentration from about 2% in Europe to 20% over India for the decade 2041–2050.
Local response to global reduction can be higher than the global temperature response, particularly for regions subjected to rapid changes. Hence, mitigation of rapid warming in the Arctic has been subject to an increasing number of studies (Sand et al. , 2013b, 2016; Jiao et al. , 2014; AMAP, 2015a, b; Mahmood et al. , 2016; Christensen et al. , 2019). Considering maximum technically feasible reductions (MTFR) for methane globally and an idealized strategy reducing key global anthropogenic sources of BC (about 80% reduction by 2030 and sustained thereafter) and precursors of ozone was estimated to jointly bring a reduction of Arctic warming, averaged over the 2041–2050 period, between 0.2°C and 0.6°C (AMAP, 2015a; Sand et al., 2016). Stohl et al. (2015) have estimated that a global SLCF mitigation strategy (excluding further reduction of SO2) would lead to about twice as high a temperature reduction (–0.44 (–0.39 to –0.49) °C) in the Arctic than the global response to such mitigation.
While there is robust evidence that air-quality policies resulting in reductions of aerosols and ozone can be beneficial for human health but can lead to ‘disbenefits’ for near-term climate change, the existence of such trade-offs in response to climate change mitigation policies is less certain (Shindell and Smith, 2019). Recent studies show that very ambitious but plausible gradual phasing out of fossil fuels in 1.5°C-compatible pathways with little or no overshoot, lead to a near-term future warming of less than 0.1°C, when considering associated emissions reduction of both warming and cooling species. This suggests that there may not be a strong conflict, at least at the global scale, between climate and air-quality benefits in the case of a worldwide transition to clean energy (Shindell and Smith, 2019; Smith et al., 2019). However, at the regional scale, the changes in spatially variable emissions and abundance changes might result in different responses, including implications for precipitation and monsoons (Chapter 8), especially over Southern Asia (e.g., Wilcox et al., 2020).
Decarbonization of energy supply and end-use sectors is among key pillars of any ambitious climate change mitigation strategy and it would result in improved air quality owing to associated reduction of co-emitted SLCF emissions (e.g., McCollum et al., 2013; Rogelj et al., 2014b; Braspenning Radu et al., 2016; Rao et al., 2016; Stechow et al., 2016; Lelieveld et al., 2019; Shindell and Smith, 2019). Regional studies (Lee et al., 2016; Shindell et al., 2016; Chen et al., 2018; Li et al., 2018), where significant CO2 reductions were assumed for 2030 and 2050, show consistently reduced of PM2.5 and ozone concentrations resulting in important health benefits. However, these improvements are not sufficient to bring PM2.5 levels in agreement with the WHO air-quality guidelines in several regions. Amann et al. (2020) and UNEP (2019) highlight that only the combination of strong air-quality, development and climate policies, including societal transformations, could pave the way towards the achievement of such a target at a regional and global level.
At a global level, Rao et al. (2016) showed that climate policies, compatible with Copenhagen pledges and a long-term CO2 target of 450 ppm, result in important air-quality benefits, reducing the share of the global population exposed to PM2.5 levels above the WHO Tier 1 standard (35 µg m–3) in 2030 from 21% to 5%. The impacts are similar to a strong air-quality policy but still leave large parts of population, especially in Asia and Africa, exposed to levels well above the WHO air quality guideline level of 10 µg m–3. The latter can be partly alleviated by combining such climate policy with strong air-quality policy. Shindell et al. (2018) analysed more ambitious climate change mitigation scenarios than Rao et al. (2016) and highlighted the opportunities to improve air quality and avert societal effects associated with warmer climate by accelerated decarbonization strategies. Most climate change mitigation strategies compatible with limiting global warming to well below 2°C rely on future negative CO2 emissions postponing immediate reduction. Alternatively, a faster decarbonization could allow the achievement of a 2°C goal without negative CO2 emissions and, with currently known and effectively applied emissions-control technologies, this would also have immediate and significant air-quality benefits, reducing premature deaths worldwide (Shindell et al., 2018). For a 2°C-compatible pathway, Vandyck et al. (2018) estimated 5% and 15% reduction in premature mortality due to PM2.5 in 2030 and 2050, respectively, compared to reference scenarios.
There is robust evidence that reducing atmospheric methane will benefit climate and improve air quality through near-surface ozone reduction (Fiore et al., 2015; Shindell et al., 2017a) and wide agreement that strategies reducing methane offer larger (and less uncertain) climate benefits than policies addressing BC (e.g., Smith andMizrahi, 2013; Rogelj et al., 2014b, 2018b; Stohl et al., 2015; Christensen et al., 2019; Shindell and Smith, 2019). SR1.5 (Rogelj et al., 2018b) highlighted the importance of methane mitigation in limiting warming to 1.5ºC in addition to net zero CO2 emissions by 2050. Implementation of the identified maximum technically feasible reductions (MTFR) potential for methane globally, estimated at nearly 50% reduction (or 205 Tg CH4 in 2050) of anthropogenic emissions from the baseline, would lead to a reduction in warming, calculated as the differences between the baseline and MTFR scenario, for the 2036–2050 period of about 0.20°C ± 0.02°C globally (AMAP, 2015b). Plausible levels of methane mitigation, achieved with proven technologies, can increase the feasibility of achieving the Paris Agreement goal through slightly slowing down the pace of CO2 reductions (but not changing the final CO2 reduction goal) while this benefit is enhanced by the indirect effects of methane mitigation on ozone levels (Collins et al., 2018). Adressing methane mitigation appears even more important in view of recently observed growth in atmospheric concentrations that is linked to increasing anthropogenic emissions (Section 5.2.2).
Neither ambitious climate change policy nor air-quality abatement policy can automatically yield co-benefits without integrated policies aimed at co-beneficial solutions (Zusman et al., 2013; Schmale et al., 2014a; Melamed et al., 2016), particularly in the energy generation and transport sectors (Rao et al. , 2013; Thompson et al. , 2016; Shindell et al. , 2018; Vandyck et al. , 2018). Integrated policies are necessary to yield multiple benefits of mitigating climate change, improving air quality, protecting human health and achieving several SDGs.
Box 6.2 | SLCF Mitigation and Sustainable Development Goals (SDG) Opportunities
Striving to achieve air-quality and climate targets will bring significant SLCF reductions. These reductions contribute first and foremost to the attainment of SDGs targeting improved human health and sustainable cities (SDGs 3 and 11), specifically related to PM exposure (goals 3.9 and 11.6; Lelieveld, 2017; Amann et al., 2020), but also access to affordable and clean energy, responsible consumption and production, and climate, as well as reducing nutrient losses and consequently protecting biodiversity (SDG 7, 12, 13, 14 and 15; UNEP, 2019; Amann et al., 2020). Furthermore, declining SLCF emissions will result in reduced crop losses (SDG 2; zero hunger) due to decrease of ozone exposure (Feng and Kobayashi, 2009; Ainsworth et al., 2012; Emberson et al., 2018).
However, the design of suitable policies addressing these SDGs can be difficult because of the complexity of linking emissions to impacts on human health, ecosystems, equity, infrastructure and costs. Beyond the fact that several species are co-emitted, interlinkage between species, such as through atmospheric chemistry, can weaken the benefit of emissions reduction efforts. An illustration lies in the recent (2013–2017) reduction of aerosols over China (Silver et al., 2018; Zheng et al., 2018b) resulting from the strategy to improve air quality (‘Clean Air Action’); this has successfully reduced the level of PM2.54 but has led to a concurrent increase in surface ozone, partly due to declining heterogeneous interactions of ozone precursors with aerosols (K. Li et al., 2019; Yu et al., 2019). This side effect on ozone has been addressed since then by amending the legislation to target NMVOC sources, especially solvent use. Complex interactions between anthropogenic and biogenic volatile compounds are also at play and reduction of certain SLCFs could possibly promote new particle formation from organic vapours (e.g., Lehtipalo et al., 2018). Finally, a recent example of this complexity is the mixed effects on ozone pollution induced by NOx decrease during the COVID-19 pandemic (Cross-Chapter Box 6.1). Thus, the climate and air pollution effects of policies depend strongly on the choice of regulated compounds and the degree of reduction. Such policies have to be informed by strong science support, including for example multi-model analyses such as HTAP (UNECE, 2010) and AMAP (AMAP, 2015a, b), based on global and regional CCMs. This is essential to capture the complexity and inform the policy development process.
In addition, pursuing SDG objectives, apparently decoupled from air pollution, such as improved waste management, access to clean energy, or improved agricultural practices, would also stimulate and lead to mitigation of SLCFs. Amann et al. (2020) show that a global strategy to achieve the WHO air quality guidelines, cannot only rely on air pollution control but also on a combination of SDG-aligned policies. Such actions would include energy efficiency improvements, increased use of renewables, reduction of methane from waste management and agriculture, and CO2 and methane due to lower fossil fuel consumption, resulting in climate co-benefits. Consideration of SDGs including local air-quality co-benefits, creates an opportunity to support and gain acceptance for ambitious climate change mitigation (Jakob and Steckel, 2016; Stechow et al., 2016; Vandyck et al., 2018). Such near-term policies targeting SDGs and air quality would enable longer-term transformations necessary to achieve climate goals (Chapter 17, WGIII).
In summary, there is high confidence that effective decarbonization strategies could lead to air-quality improvements but are not sufficient to achieve, in the near term, air-quality WHO guideline values set for fine particulate matter, especially in parts of Asia and in some highly polluted regions. Additional policies (e.g., access to clean energy, waste management) envisaged to attain SDGs bring complementary SLCF reduction (high confidence). Sustained methane mitigation, wherever it occurs, stands out as an option that combines near- and long-term gains on surface temperature (high confidence) and leads to an air pollution benefit by reducing ozone levels globally (high confidence) .
Cross-Chapter Box 6.1 | Implications of COVID-19 Restrictions for Emissions, Air Quality and Climate
Coordinators: Astrid Kiendler-Scharr (Germany/Austria), John C. Fyfe (Canada)
Contributors: Josep G. Canadell (Australia), Sergio Henrique Faria (Spain/Brazil), Piers Forster (UK), Sandro Fuzzi (Italy), Nathan P. Gillett (Canada), Christopher Jones (UK), Zbigniew Klimont (Austria/Poland), Svitlana Krakovska (Ukraine), Prabir Patra (Japan/India), Joeri Rogelj (Austria/Belgium), Bjørn Samset (Norway), Sophie Szopa (France), Izuru Takayabu (Japan), Hua Zhang (China)
In response to the outbreak of COVID-19 (officially the severe acute respiratory syndrome–coronavirus 2 or SARS-CoV-2), which was declared a pandemic on March 11 2020 by the World Health Organization (WHO), regulations were imposed by many countries to contain the spread of COVID-19. Restrictions were implemented on the movement of people, such as closing borders or requiring the majority of population to stay at home, for periods of several months. This Cross-Chapter Box assesses the influence of the COVID-19 containment on short-lived climate forcers (SLCFs) and long-lived greenhouse gases (LLGHGs), and related implications for the climate. Note that this assessment was developed late in the AR6 WGI process and is based on the available emerging literature.
Emissions
Global fossil CO2 emissions are estimated to have declined by 7% (medium confidence) in 2020 compared to 2019 emissions, with estimates ranging from 5.8% to 13.0% based on various combinations of data on energy production and consumption, economic activity and proxy activity data for emissions and their drivers (Forster et al. , 2020; Friedlingstein et al. , 2020; Le Quéré et al. , 2020; Liu et al. , 2020). However, the concentration of atmospheric CO2 continued to grow in 2020 compared to previous years (Dlugokencky and Tans, 2021). Given the large natural interannual variability of CO2 (Section 5.2.1), and the small expected impact of emissions in the CO2 growth rate, there were no observed changes in CO2 concentration that could be attributed to COVID-19 containment (medium confidence) (Chevallier et al., 2020; Tohjima et al., 2020).
Global daily CO2 emissions from fossil fuel sources had a maximum decline of 17% in early April, compared with the mean 2019 levels, and coinciding with the global peak pandemic lockdown (Le Quéré et al., 2020). The reductions in CO2 emissions in 2020 were dominated by the drop in emissions from surface transport followed, in order of absolute emissions reductions, by industry, power and aviation (Le Quéré et al., 2020; Liu et al., 2020). Residential emissions showed little change (Liu et al., 2020) or rose slightly (Forster et al., 2020; Le Quéré et al., 2020). Aviation had the biggest relative drop in activity. CO2 emissions due to land use (based on early and uncertain evidence on deforestation and forest fires) were higher than average in 2020 (Amador-Jiménez et al., 2020).
Using similar methodologies, Forster et al. (2020) assembled activity data and emissions estimates for other greenhouse gases and aerosols and their precursors. Anthropogenic NOx emissions, which are largely from the transport sector, are estimated to have decreased by a maximum of 35% in April (medium confidence). Species whose emissions are dominated by other sectors, such as methane and NH3 from agriculture, saw smaller reductions.
Abundances and air quality
Owing to the short atmospheric lifetimes of SLCFs relevant to air quality, changes in their concentrations were detected within a few days after lockdowns had been implemented (e.g., Bauwens et al. , 2020; Venter et al. , 2020; Gkatzelis et al. , 2021; Shi et al. , 2021). The COVID-19-driven economic slowdown has illustrated how complex the relationship is between emissions and air pollutant concentrations due to non-linearity in the atmospheric chemistry leading to secondary compound formation (Section 6.1, Box 6.1; Kroll et al., 2020).
Several studies have examined the effect of COVID-19 containment on air quality, showing that multi-year datasets with proper statistical/modelling analysis are required to discriminate the effects of meteorology from that of emissions reduction (Dhaka et al. , 2020; L. Li et al. , 2020; Wang et al. , 2020; Zhao et al. , 2020b). Accounting for meteorological influences and with an increasing stringency index, the median observed change in NO2 decreased from –13% to –48%, and in PM2.5 decreased from –10% to –33%, whereas the median change in ozone increased from 0% to 4% (Gkatzelis et al., 2021). The latter can be explained by the decrease of NO emissions that titrate ozone in specific highly polluted areas, leading to the observed increase in surface ozone concentration in cities (Le et al. , 2020; Sicard et al. , 2020; Huang et al. , 2021).
The temporary decrease of PM2.5 concentrations should be put in perspective of the sustained reduction (estimated at 30–70%), which could be achieved by implementing policies addressing air quality and climate change (Section 6.6.3). Such sustained reductions can lead to multiple benefits and simultaneously achieve several SDGs (Section 6.6.3). These policies would also result in reduction of ground-level ozone by up to 20% (Section 6.7.1.3).
Except for ozone, temporary improvement of air quality during lockdown periods was observed in mostregions of the world (high confidence), resulting from a combination of interannual meteorological variability and the impact of COVID-19 containment measures (high confidence). Estimated air pollution reductions associated with lockdown periods are lower than what can be expected from integrated mitigation policy leading to lasting reductions (medium confidence).
Radiative forcings
COVID-19-related emissions changes primarily exerted effective radiative forcing (ERF) through reduced emissions rates of CO2 and methane, altered abundance of SLCFs, notably ozone, NO2 and aerosols, and through other changes in anthropogenic activities, notably a reduction in the formation of aviation-induced cirrus clouds.
Forster et al. (2020) combined the FaIR emulator (Cross-Chapter Box 7.1) with emissions changes for a range of species, relative to a continuation of Nationally Determined Contributions (Rogelj et al., 2017). They found a negative ERF from avoided CO2 emissions that strengthens through 2020 to –0.01 W m–2. During the spring lockdown, they found a peak positive ERF of 0.1 W m–2 from loss of aerosol-induced cooling, and a peak negative ERF of –0.04 W m–2 from reductions in tropospheric ozone (from reduced photochemical production via NOx). Overall, they estimated a net ERF of +0.05 W m–2 for spring 2020, declining to +0.025 W m–2 by the end of the year.
Cross-Chapter Box 6.1
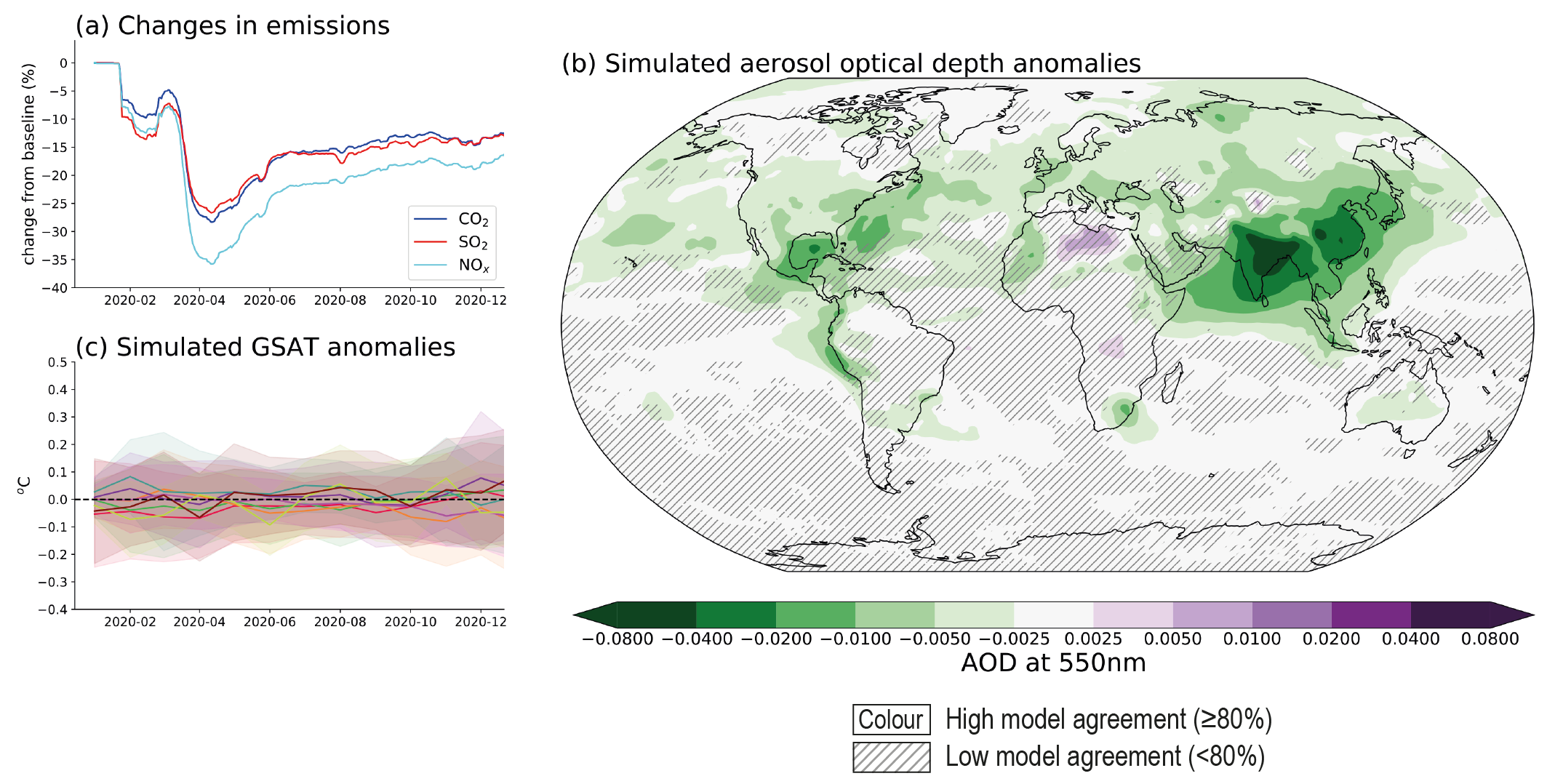
Cross-Chapter Box 6.1, Figure 1 | Emissions reductions and their effect on aerosols and climate in response to COVID-19. Estimated reductions in emissions of CO2, SO2 and NOx are shown in panel (a) based on reconstructions using activity data (updated from Forster et al., 2020). Eight Earth system models (ESMs) performed multiple ensemble simulations of the response to COVID-19 emissions reductions forced with these assumed emissions reductions up until August 2020 followed by a constant continuation near the August value to the end of 2020. Emissions reductions were applied relative to the SSP2-4.5 scenario. Panel (b) shows ESM-simulated AOD at 550nm (only seven models reported this variable). Panel (c) shows ESM-simulated GSAT anomalies during 2020; curves denote the ensemble mean result for each model with shading used for ±1 standard deviation for each model. ESM data from these simulations (‘ssp245-covid’) is archived on the Earth System Grid CMIP6 database. Uncertainty is represented using the simple approach: no overlay indicates regions with high model agreement, where ≥80% of models agree on sign of change; diagonal lines indicate regions with low model agreement, where <80% of models agree on sign of change. For more information on the simple approach, please refer to the Cross-Chapter Box Atlas.1.
Gettelman et al. (2021) extended Forster et al.’s (2020) results using two ESMs, and found a spring peak aerosol-induced ERF ranging from 0.12 to 0.3 W m–2, depending on the aerosol parametrization. They also estimated an ERF of –0.04 W m–2 from loss of contrail warming. Overall, they report a peak ERF of 0.04 to 0.2 W m–2, and a subsequent decline to around half the peak value. Two independent ESM studies Weber et al. (2020) and Yang et al. (2020) found consistent results in time evolution and component contributions but included fewer forcing components.
The available studies are in broad agreement on the sign and magnitude of contributions to ERF from COVID-19-related emissions changes during 2020. The range in peak global mean ERF in spring 2020 was [0.025 to 0.2] W m–2 (medium confidence), composed of a positive forcing from aerosol–climate interactions of [0.1 to 0.3] W m–2, and negative forcings from CO2 (–0.01 W m–2), NOx (–0.04 W m–2) and contrail cirrus (–0.04 W m–2) (limited evidence, medium agreement). By the end of 2020, the ERF was at half the peak value (medium confidence).
Climate responses
Changes in atmospheric composition due to COVID-19 emissions reductions are not thought to have caused a detectable change in global temperature or rainfall in 2020 (high confidence). A large ensemble of Earth system model (ESM) simulations show an ensemble average reduction in Aerosol Optical Depth (AOD) in some regions, notably Eastern and Southern Asia (Fyfe et al., 2021). This result is supported by observational studies finding decreases in optical depth in 2020 (Gkatzelis et al., 2021; Ming et al., 2021; van Heerwaarden et al., 2021), which may have contributed to observed increases in solar irradiance (van Heerwaarden et al., 2021) or solar clear-sky reflection (Ming et al., 2021).
Cross-Chapter Box 6.1
Model simulations of the response to COVID-19 emissions reductions indicate a small warming of global surface air temperature (GSAT) due to a decrease in sulphate aerosols (Forster et al., 2020; Fyfe et al., 2021), balanced by cooling due to an ozone decrease (Forster et al., 2020; Weber et al., 2020), black carbon decrease (Weber et al., 2020) and CO2 decrease. It is noted that observational studies report little SO2 change, at least locally near the surface (Shi et al., 2021), and do not correlate with emissions inventory-based changes (Gkatzelis et al., 2021). One study suggests a small net warming while another using idealized simulations suggests a small cooling (Weber et al., 2020). Simulated GSAT and rainfall changes are unlikely to be detectable in observations (high confidence) (Samset et al., 2020; Fyfe et al., 2021). Multi-model ESM simulations based on a realistic COVID-19 containment forcing scenario (Forster et al., 2020) indicate a model mean reduction in regional AOD but no discernible response in GSAT (Figure 1, Cross-Chapter Box 6.1).
6.7 Future Projections of Atmospheric Composition and Climate Response in SSP Scenarios
This section assesses the 21st-century projections of SLCF emissions, abundances and responses in terms of climate and air quality following the SSPs (Chapter 1, Section 1.6.1.3 and Cross-Chapter Box 1.5; Riahi et al., 2017; Gidden et al., 2019). The future evolution of atmospheric abundances and the resulting climate and AQ responses is driven mainly by anthropogenic emissions and by natural emissions modulated by chemical, physical and biological processes as discussed in Sections 6.2 and 6.3. Like the RCP scenarios used in AR5, the SSP emissions scenarios consider only direct anthropogenic (including biomass burning) emissions and do not project natural emissions changes due to climate or land-use changes; ESMs intrinsically consider these biogeochemical feedbacks to varying degrees (Section 6.4.5). We rely on future projections based on CMIP6 ESMs with comprehensive representation of chemistry, aerosol microphysics and biospheric processes that participated in the ScenarioMIP (O’Neill et al., 2016) and AerChemMIP (Collins et al., 2017). However, due to the high computational costs of running coupled ESMs, they cannot be used for quantifying the contributions from individual species, regions and sectors, and across the scenarios. Therefore, reduced complexity models (Box 1.3 and Cross-Chapter Box 7.1), which represent chemistry and complex ESM interactions in parametrized forms updated since the AR5, are also applied here.
6.7.1 Projections of Emissions and Atmospheric Abundances
6.7.1.1 SLCF Emissions and atmospheric abundances
The trajectory of future SLCF emissions is driven by the evolution of socio-economic drivers described in Section 1.6.1.1 but dedicated, SSP-specific, air pollution policy storylines can change the regional and global trends (Rao et al., 2017). Additionally, assumptions about urbanization (Jiang and O’Neill, 2017) will affect the spatial distribution of emissions and consequently air quality.
Growing urbanization worldwide has strongly modified the spatial distribution and intensity of SLCF emissions. The effect and extent of urbanization on air pollution and other emissions species are captured within Integrated Assessment Models (IAMs) at varying levels of complexity. In most cases, models use a combination of proxies and assumptions of end-use efficiency and technological improvement assumptions to estimate emissions arising from rural-to-urban migration and population growth within cities, utilizing quantifications of urbanization for the SSPs (Jiang and O’Neill, 2017). In addition, spatial patterns of future rural and urban population growth, migration, and decline have been quantified for the SSPs using a gravity model (Jiang and O’Neill, 2017). However, linking these spatial patterns with IAM regional emissions pathways is still an ongoing area of study and has not yet been represented in spatial emissions estimates provided by IAMs (Riahi et al., 2017; Gidden et al., 2019; Feng et al., 2020). As described in Feng et al. (2020), spatial emissions estimates derived for CMIP6 are largely a product of existing spatial patterns of population, but do not vary dynamically in future patterns. To the extent urbanization is accounted for in gridded emissions, IAM native region resolution (varying, for example, from 11 world regions to more than 30, depending on the model) provides urbanization-based dynamics. Despite the interest of studying the effect of well-planned, densely populated urban centres, which can help to maximize the benefits of agglomeration, by providing proximity to infrastructure and services, the opportunity for energy saving, and providing a frame for air-quality control, IAM realizations of SSPs are not sufficient to assess this effect. The opportunities and risks associated with this rapid urbanization for SLCF emissions and air quality are analysed in the Chapter 6 of the WGII report and Chapter 8 of the WGIII report.
All the RCP trajectories started in 2005 and relied on the assumption that economic growth will bring rapid strengthening of air pollution legislation, effectively reducing emissions of non-methane SLCFs (e.g., Chuwah et al., 2013). While in the long-term such trends are expected if more ambitious air pollution control goes on par with economic growth. The near-term developments, however, might be much more diverse across regions and species, as has been observed in the last three decades (Amann et al., 2013; Rafaj et al., 2014; Rafaj and Amann, 2018; Ru et al., 2018), especially in several fast-growing economies, leading to the difference between CMIP6 historical estimates for the post-2000 period (Hoesly et al., 2018) and those used in RCPs (Figure 6.18). Since several SLCFs are also air pollutants, the narrow range of the RCP emissions trajectories in the future allowed for only limited analysis of near-future air quality (Amann et al. , 2013; Chuwah et al. , 2013; von Schneidemesser et al. , 2015). However, the range of storylines in the SSPs lead to a wider range of assumed pollution-control policies in the SSPs (Rao et al., 2017; Riahi et al., 2017). In SSP1 and SSP5, strong air-quality policies are assumed to minimize the adverse effects of pollution on population and ecosystems. In SSP2, a medium pollution control, with lower than current policy targets, is considered. Only weak, regionally varied, air pollution policies are applied in the SSP3 and SSP4. Additional climate policies introduced to reach defined radiative forcing targets will also affect SLCF emissions. The SSP SLCF-emissions trajectories (Rao et al., 2017; Gidden et al., 2019) assume a long-term coupling of economic growth and specific emissions indicators, such as sectoral emission densities. The pace of change varies across regions and SSPs resulting in a wider range of future air pollutants evolution (Figure 6.18), reflecting the differences in assumed levels of air pollution controls across regions (Figure 6.19). At the end of the century, the range across the SSPs is about four times that of RCPs for SO2 and NOx, two to four times for BC and NMVOCs, and up to three times for CO and OC, while indicating a slightly smaller range than RCPs for methane (Figure 6.18). The originally developed SSP scenarios (Rao et al., 2017) have been harmonized with the CMIP6 historical emissions (Hoesly et al., 2018) and include updated SO2 emissions to account for the recent decline in China (Gidden et al., 2019).
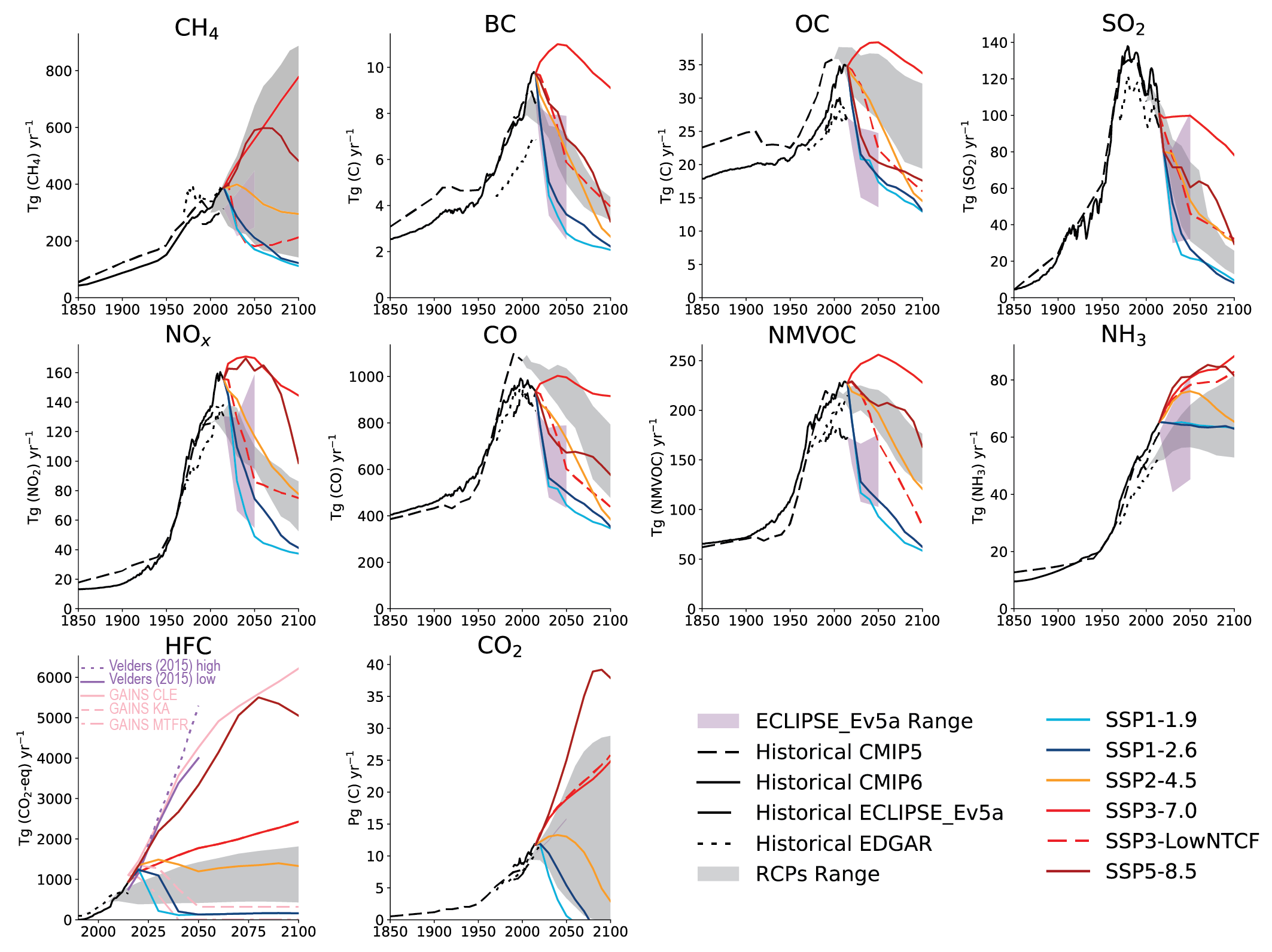 Figure 6.18 | Global anthropogenic and biomass burning short-lived climate forcer (SLCF) and CO2 emissions from 1850 to 2100 and HFC emissions from 1990 to 2100. Emissions for the Coupled Model Intercomparison Project Phase 6 (CMIP6) for the period 1850–2014 are based on Hoesly et al. (2018) and van Marle et al. (2017); emissions for CMIP5 for the period 1850–2005 are from Lamarque et al. (2010); CO2 emissions are from EDGAR database (Crippa et al., 2020); methane (CH4) and HFCs are from (Crippa et al., 2019); and air pollutants are from EC-JRC / PBL (2020), Höglund-Isaksson (2012) and Klimont et al. (2017a) for ECLIPSE. Projections originate from the Shared Socio-Economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019); Representative Concentration Pathway (RCP) database (van Vuuren et al., 2011); GAINS (CLE – current legislation baseline, KA – Kigali Amendment, MTFR – maximum technical mitigation potential) for HFCs (Purohit et al., 2020; Velders et al. 2015); and, ECLIPSE (Stohl et al., 2015). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.18 | Global anthropogenic and biomass burning short-lived climate forcer (SLCF) and CO2 emissions from 1850 to 2100 and HFC emissions from 1990 to 2100. Emissions for the Coupled Model Intercomparison Project Phase 6 (CMIP6) for the period 1850–2014 are based on Hoesly et al. (2018) and van Marle et al. (2017); emissions for CMIP5 for the period 1850–2005 are from Lamarque et al. (2010); CO2 emissions are from EDGAR database (Crippa et al., 2020); methane (CH4) and HFCs are from (Crippa et al., 2019); and air pollutants are from EC-JRC / PBL (2020), Höglund-Isaksson (2012) and Klimont et al. (2017a) for ECLIPSE. Projections originate from the Shared Socio-Economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019); Representative Concentration Pathway (RCP) database (van Vuuren et al., 2011); GAINS (CLE – current legislation baseline, KA – Kigali Amendment, MTFR – maximum technical mitigation potential) for HFCs (Purohit et al., 2020; Velders et al. 2015); and, ECLIPSE (Stohl et al., 2015). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). All SSP scenarios (Figure 6.18), except SSP3-7.0, project decline in global total emissions for all SLCFs by the end of the 21st century, except for ammonia and for HFCs (more on HFCs below). Similar to RCPs, ammonia emissions continue to increase in most SSPs, except SSP1 and SSP2, accounting for the expected growth in food demand and a general lack of effective policies targeting agricultural emissions. Additionally, mitigation potential for NH3 is generally smaller than for other species owing to fugitive and widely distributed sources (Pinder et al., 2007; Klimont and Winiwarter, 2015; Mohankumar Sajeev et al., 2018; Sajeev et al., 2018). Most significant changes of SLCF emissions in the near and long-term compared to the present day are expected for SO2. This is due to ever more stringent (and enforced) legislation in China’s power sector, extended recently to industrial sources (Zheng et al., 2018b; Tong et al., 2020), declining coal use in most SSPs, recently announced stricter emissions limits for the power sector in India, and reduction of the sulphur content of oil fuel used in international shipping from 2020 (IMO, 2016). For the lower forcing targets (e.g., SSP1-2.6), the SO2 trajectories are similar to the RCPs resulting in over 50–90% decline by 2050 and 2100, respectively, while for the scenarios with no climate policies, the SSPs show a large spread even at the end of the century.
Until the mid-21st century, SSP3-7.0 and SSP5-8.5 scenarios project no reduction in NOx emissions at the global level with decline in most OECD countries and Eastern Asia, driven by existing legislation in power, industry and transportation (e.g., Tong et al., 2020), and continued increase in the rest of the world (Figures 6.18 and 6.19). Towards the end of the century, similar trends continue in SSP3-7.0 while emissions in SSP5 decline strongly owing to faster technological progress and stronger air-quality action (Rao et al., 2017; Riahi et al., 2017). By 2100, the ‘Regional Rivalry’ (SSP3) scenario emissions of NOx (and most other SLCFs, except ammonia) are typically twice as high as the next highest SSP projection, both at the global (Figure 6.18) and regional levels (Figure 6.19). In emissions pathways consistent with Paris Agreement goals (SSP1-1.9 or SSP1-2.6; Section 1.6.1), NOx drops, compared to 2015, by 50% in SSP1-2.6 and by 65% in SSP1-1.9 by 2050, is reduced by about 70% by 2100, resulting in global emissions levels comparable to the 1950s and below the RCP range. In these pathways considering strong climate change mitigation, similar reductions are projected at the regional level, except in Africa (less than 50% decline) due to its high share of biomass emissions as well as strong growth in population and fossil fuel use. The trends in anthropogenic and biomass-burning emissions for other ozone precursors (NMVOC, CO) are similar to that of NOx.
An additional scenario, based on the SSP3-7.0, has been designed specifically to assess the effect of a strong SLCF emissions abatement and is called SSP3-7.0-lowNTCF in the literature (Collins et al., 2017; Gidden et al., 2019). It has been applied in the modelling studies (e.g., AerChemMIP) with or without consideration of additional methane reduction and we refer here to these scenarios, respectively, as SSP3-7.0-lowSLCF-lowCH4 or SSP3-7.0-lowSLCF-highCH4. In these scenarios, aerosols, their precursors, and non-methane tropospheric ozone precursors are mitigated by applying the same emissions factors as in SSP1-1.9.
For global methane emissions, the range is similar for SSPs and RCPs over the entire century (Figure 6.18), with highest projections in SSP3-7.0 (slightly below RCP8.5) estimating doubling of the current emissions and a reduction of about 75% by 2100 in scenarios consistent with 1.5°C to 2°C targets; similar to RCP2.6. At the regional level, the evolution of methane emissions in climate change mitigation scenarios is comparable to RCPs but there are significant differences for some regions with respect to high CO2 emissions scenarios. In particular, projections for Eastern Asia differ significantly, the highest SSP3-7.0 is about half of the highest RCP by 2100 (Figure 6.19), which is due to much lower projections of coal use in China driven largely by efforts during the last decade to combat poor air quality. At the same time, the SSP scenarios without climate change mitigation project faster growth in methane emissions in Africa, the Middle East and Latin America (Figure 6.19) driven by developments in agriculture, the oil and gas sectors, and, especially in Africa, waste management. There are significant differences in the assessment and feasibility of rapid methane mitigation. Höglund-Isaksson et al. (2020) review most recent studies and assess feasibility of rapid widespread mitigation, concluding that significant (over 50%) reductions are attainable but the feasibility of such reductions could be constrained in the short term due to locked capital. This might have implications for near-term evolution assumed in, for example, SSP1-1.9 or SSP3-lowSLCF-lowCH4, where emissions drop very quickly due to fast decarbonization and reductions in agriculture. Such high reduction potential in agriculture has been also assumed in other studies (Lucas et al., 2007; Harmsen et al., 2020a) but is questioned by Höglund-Isaksson et al. (2020) who indicate that widespread implementation (within decades) of policies bringing about institutional and behavioural changes would be important for transition towards very low methane emissions from livestock production.
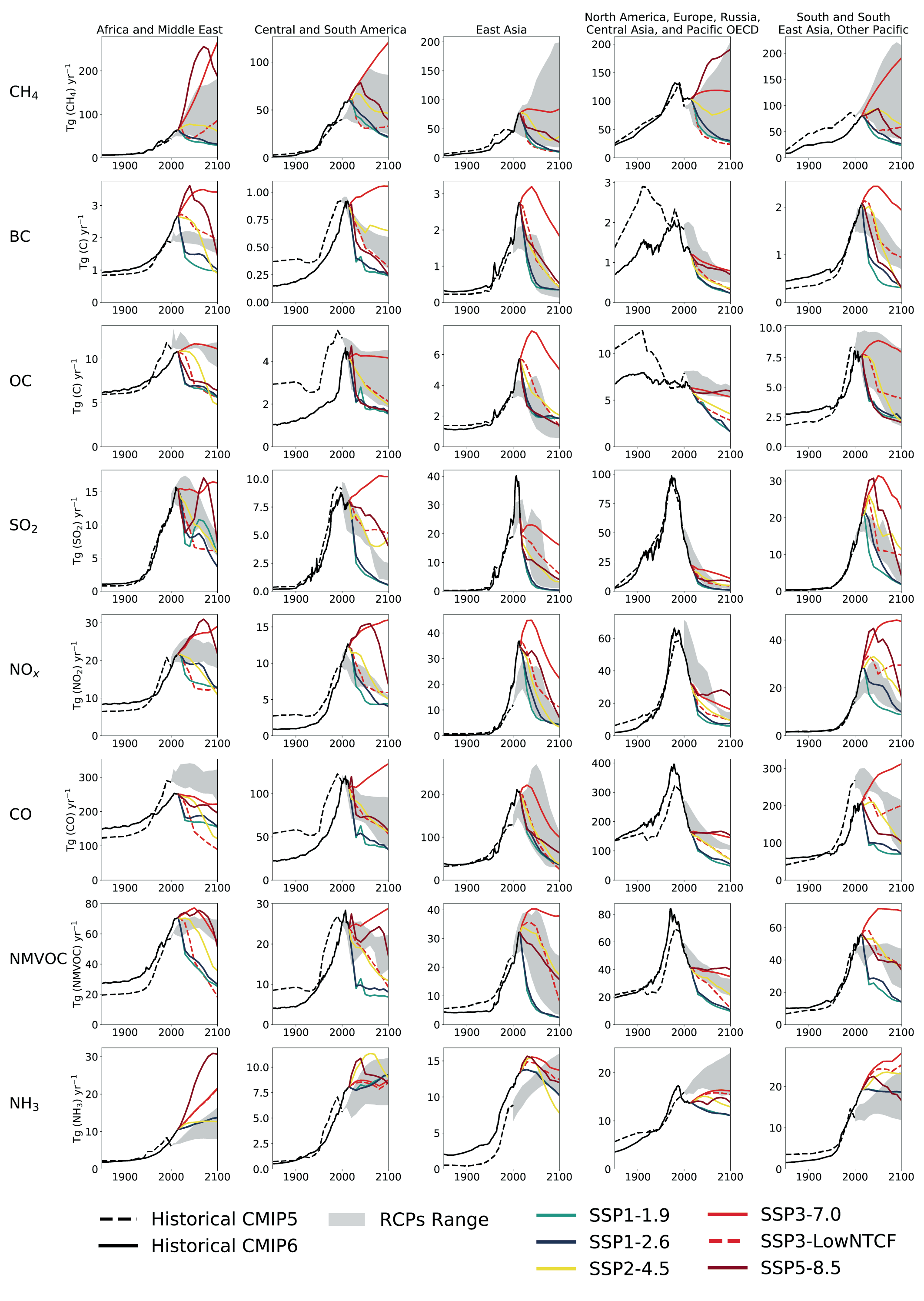 Figure 6.19 | Regional anthropogenic and biomass burning short-lived climate forcer (SLCF) emissions from 1850 to 2100. Emissions for the Coupled Model Intercomparison Project Phase 6 (CMIP6) for the period 1850–2014 are based on Hoesly et al. (2018) and van Marle et al. (2017) and emissions for CMIP5 for the period 1850–2005 are from Lamarque et al. (2010). Projections originate from the Shared Socio- economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019) and Representative Concentration Pathway (RCP) database (van Vuuren et al., 2011). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.19 | Regional anthropogenic and biomass burning short-lived climate forcer (SLCF) emissions from 1850 to 2100. Emissions for the Coupled Model Intercomparison Project Phase 6 (CMIP6) for the period 1850–2014 are based on Hoesly et al. (2018) and van Marle et al. (2017) and emissions for CMIP5 for the period 1850–2005 are from Lamarque et al. (2010). Projections originate from the Shared Socio- economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019) and Representative Concentration Pathway (RCP) database (van Vuuren et al., 2011). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).Global emissions of carbonaceous aerosols are projected to decline in all SSP scenarios (Figure 6.18) except SSP3-7.0. In that scenario, which also has much higher emissions than any of the RCPs, about half of the anthropogenic BC originates from cooking and heating on solid fuels, mostly in Asia and Africa (Figure 6.19), where only limited progress in access to clean energy is achieved. Slow progress in improving waste management, high coal use in energy and industry, and no further progress in controlling diesel engines in Asia, Africa and Latin America contributes most of the remaining emissions, resulting in about 90% of anthropogenic BC emitted in the non-OECD world by 2100 in SSP3-7.0. A similar picture emerges for OC but with greater contribution of the waste management sector and biomass burning, and lower impact of transportation and industry developments. Since scenarios compliant with Paris Agreement goals (SSP1-1.9 or SSP1-2.6; Section 1.6.1) include widespread access to clean energy already by 2050, the global and regional emissions of BC decline by 70–75% by 2050 and 80% by 2100 relayive to 2015. The decline in the residential sector (about 90% by 2050 and over 95% by 2100) is accompanied by a strong reduction in transport (over 98%) and the decarbonization of the industry and energy sector. About 50% of remaining BC emissions in SSP1-1.9 or SSP1-2.6 are projected to originate from waste and open biomass burning of which open burning of waste represent a significant part. Some studies suggest this might be pessimistic as, for example, efficient waste management (consistent with SDG goals) could potentially eliminate the open burning of solid residues (Gómez-Sanabria et al., 2018), which accounts for over 30% of BC emissions in SSP1-1.9 in 2050 or 2100.
The SSP scenarios draw on the HFC projections developed by Velders et al. (2015) considering, in climate change mitigation scenarios, the provisions of the Kigali Amendment (2016) to the Montreal Protocol leading to phase-down of HFCs (Section 6.6.3.2; Papanastasiou et al., 2018). The SSP scenarios without climate change mitigation (e.g, SSP3-7.0, SSP5-8.5) show a range in HFC emissions of 3.2–5.3 GtCO2-eq yr–1 in 2050 and about 4–7.2 GtCO2-eq yr–1by 2100 while in deep climate change mitigation scenarios (SSP1-1.9 and SSP1-2.6), consistent with the 1.5–2°C targets, they are expected to drop to 0.1–0.35 GtCO2-eq yr–1 (Figure 6.18). In SSP1-1.9, the extent of reduction and its pace is more ambitious than current estimates of the effect of the fully implemented and enforced Kigali Amendment (Figure 6.18; Höglund-Isaksson et al., 2017; Purohit and Höglund-Isaksson, 2017). The best representation of the HFC emissions trajectories in the SSP framework compliant with the Kigali Amendment is the SSP1-2.6 and the baseline (including only pre-Kigali national legislation; Section 6.6.3) is best represented by SSP5-8.5 (Figure 6.18). However, since HFC emissions in SSPs were developed shortly after the Kigali Amendment had been agreed, none of these projections represents accurately the HFC emissions trajectory corresponding to the phase-out emissions levels agreed to in the Kigali Amendment (Meinshausen et al., 2020), leading to medium confidence in the assessment of the benefits of the Kigali Amendment when using SSP projections for HFCs.
The SSP SLCF trajectories reflect the effect of recent legislation and the assumed evolution thereof in the longer term, however, they do not necessarily reflect the full mitigation potential for several SLCFs, within particular SSPs (Figure 6.18), that could be achieved with air-quality- or SDG-targeted policies (Amann et al. , 2013; Rogelj et al. , 2014a; Haines et al. , 2017; Klimont et al. , 2017b; Rafaj and Amann, 2018; Shindell et al. , 2018; Tong et al. , 2020). Such policies could bring more rapid mitigation of SLCFs, independent of the climate strategy (Section 6.6.3).
The projections of future SLCF abundances typically follow their emissions trajectories except for SLCFs that are formed from precursor reactions (e.g., tropospheric ozone) or are influenced by biogeochemical feedbacks (Sections 6.2.2 and 6.4.5). According to multi-model CMIP6 simulations, total column ozone (reflecting mostly stratospheric ozone) is projected to return to 1960s values by the middle of the 21st century under the SSP2-4.5, SSP3-7.0, SSP4-3.4, SSP4-6.0 and SSP5-8.5 scenarios (Keeble et al., 2021). ESMs project increasing tropospheric ozone burden over the 2015–2100 period for the SSP3-7.0 scenario (Figure 6.4; Griffiths et al., 2021), there is, however, a large spread in the magnitude of this increase reflecting structural uncertainties associated with the model representation of processes that influence tropospheric ozone. Sources of uncertainties in SLCF-abundance projections include scenario uncertainties, or parametric and structural uncertainties in the model representation of the processes affecting simulated abundances with implications for radiative forcing and air quality. The evolution of methane abundances in SSP scenarios, for example, is derived from integrated assessment models (IAMs) which do not include the effects from biogeochemical feedbacks (e.g., climate-driven changes in wetland emissions; Meinshausen et al., 2020) introducing uncertainty.
In summary, in SSPs, in addition to the socio-economic development and climate change mitigation policies shaping the GHG emissions trajectories, the SLCF emissions trajectories are also steered by varying levels of air pollution control originating from SSP narratives and independent from climate change mitigation. Consequently, SSPs span a wider range of SLCF emissions than considered in the RCPs, better covering the diversity of future options in air pollution management and SLCF-induced climate effects (high confidence). In addition to SSP-driven emissions, the future evolution of SLCFs abundance is also sensitive to chemical and biogeochemical feedbacks involving SLCFs, particularly natural emissions, whose magnitude and sign are poorly constrained.
6.7.1.2 Future Evolution of Surface Ozone and PM Concentrations
The projection of air-quality relevant abundances (surface ozone and PM2.5) under the SSP scenarios are assessed here. Future changes in global and regional annual mean surface ozone and PM2.5 driven by the evolution of emissions as well as by climate change have been quantified by CMIP6 models analysed in AerChemMIP (Allen et al., 2020, 2021; Turnock et al., 2020).
Surface ozone increases continuously until 2050 across most regions in SSP3-7.0 and SSP5-8.5, (Turnock et al., 2020), particularly over Eastern Asia, Southern Asia, the Middle East, Africa, South East Asia and Developing Pacific, where this increase can reach and even exceed 5 ppb for annual mean averaged over land areas (Figure 6.20). After 2050, surface ozone concentrations decrease in SSP5-8.5, reaching levels below their 2005–2014 mean levels in most regions, but level off or continue to increase under SSP3-7.0. The increase in surface ozone in the SSP5-8.5 scenario occurs despite an emissions decrease of several ozone precursors because the methane emissions increase until about 2080 in the absence of climate change mitigation. Ozone decreases over all regions in response to strong emissions mitigation in SSP1-1.9 and SSP1-2.6 (Turnock et al., 2020), with decreases of 5 to 10 ppb as soon as 2030 in North America, Europe, Eurasia, Eastern Asia, the Middle East and Southern Asia in their annual means over land areas. In most regions surface ozone is reduced slightly or remains near present day values in the middle of the road scenario, SSP2-4.5. In 2100, the largest differences in surface ozone changes across the scenarios occur for the Middle East, Southern Asia and Eastern Asia with differences ranging up to 40 ppb between SSP3-7.0 and SSP1-1.9 at the end of the century. In each scenario, despite discrepancies in the magnitude of changes, especially over North America, Europe, Eurasia, Eastern Asia and Southern Asia, the models are in high agreement regarding the signs of the changes and are thus assessed as of high confidence.
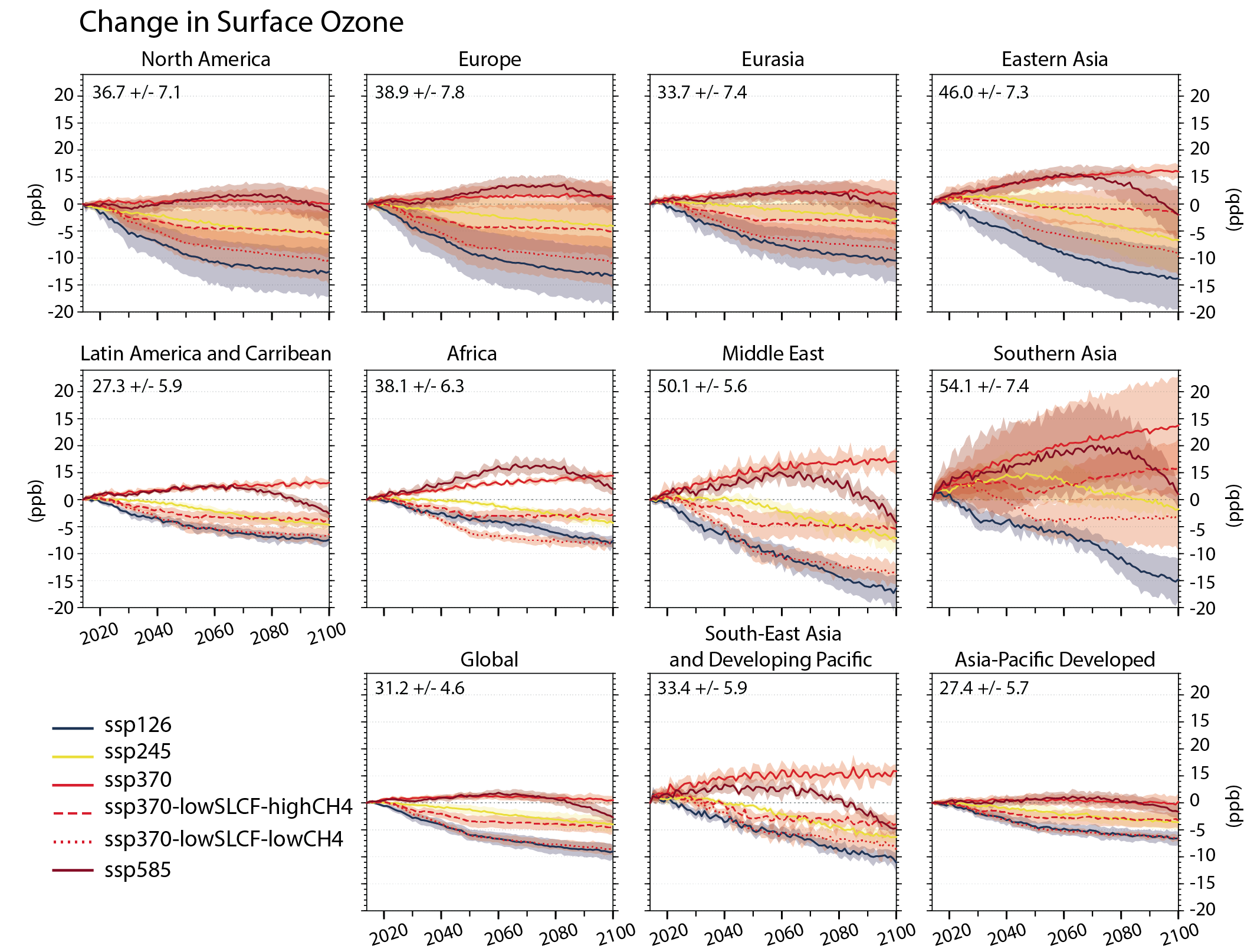 Figure 6.20 | Projected changes in regional annual mean surface ozone (O3; ppb) from 2015 to 2100 in different shared socio-economic pathways (SSPs). Each panel represents values averaged over the corresponding land area (except for ‘Global’) shown on the map in Figure 6.7. Solid coloured lines and shading indicate the multi-model mean and ±1 standard deviation across the available CMIP6 models (Turnock et al. 2020; Allen et al. 2021) for each scenario. Changes are relative to annual mean values calculated over the period 2005–2014 from the historical experiment as indicated in the top left of each regional panel along with ±1 standard deviation. For each model all available ensemble members are averaged before being used to calculate the multi-model mean. Ozone changes are also displayed in the Interactive Atlas. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.20 | Projected changes in regional annual mean surface ozone (O3; ppb) from 2015 to 2100 in different shared socio-economic pathways (SSPs). Each panel represents values averaged over the corresponding land area (except for ‘Global’) shown on the map in Figure 6.7. Solid coloured lines and shading indicate the multi-model mean and ±1 standard deviation across the available CMIP6 models (Turnock et al. 2020; Allen et al. 2021) for each scenario. Changes are relative to annual mean values calculated over the period 2005–2014 from the historical experiment as indicated in the top left of each regional panel along with ±1 standard deviation. For each model all available ensemble members are averaged before being used to calculate the multi-model mean. Ozone changes are also displayed in the Interactive Atlas. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). The strong abatement of ozone precursor emissions (except those of methane; SSP3-7.0-lowSLCF-highCH4) lead to a decrease of global average surface ozone by 15% (6 ppb) between 2015 and 2055 (Allen et al., 2020), and ozone decreases in all regions except Southern Asia. However, this decrease is twice as large when methane emissions are abated simultaneously (SSP3-7.0-lowSLCF-lowCH4), underlying the importance of methane emissions reduction as an important lever to reduce ozone pollution (high confidence) (Section 6.6.4).
A decrease in surface PM2.5 concentrations is estimated for SSP1-1.9, SSP1-2.6 and SSP2-4.5 (Turnock et al., 2020) (Figure 6.21). A decrease in PM2.5, is also projected in SSP5-8.5, which does not consider any climate change mitigation but has a strong air pollution control. The decrease is largest in the regions with the highest 2005–2014 mean concentrations (the Middle East, Southern Asia and Eastern Asia). Under the SSP3-7.0 scenario, PM2.5 is predicted to increase or remain at near present-day values across Asia; regions where present-day concentrations are currently the highest. There is large model spread over regions with large natural aerosol sources, for example, in North Africa, where dust sources are important. The mitigation of non-methane SLCFs in the SSP3-7.0-lowSLCF-highCH4 scenario is predicted to reduce PM2.54 by 25% (in 2055, relative to the SSP3-7.0 scenario) over global land surface areas (Allen et al., 2020).
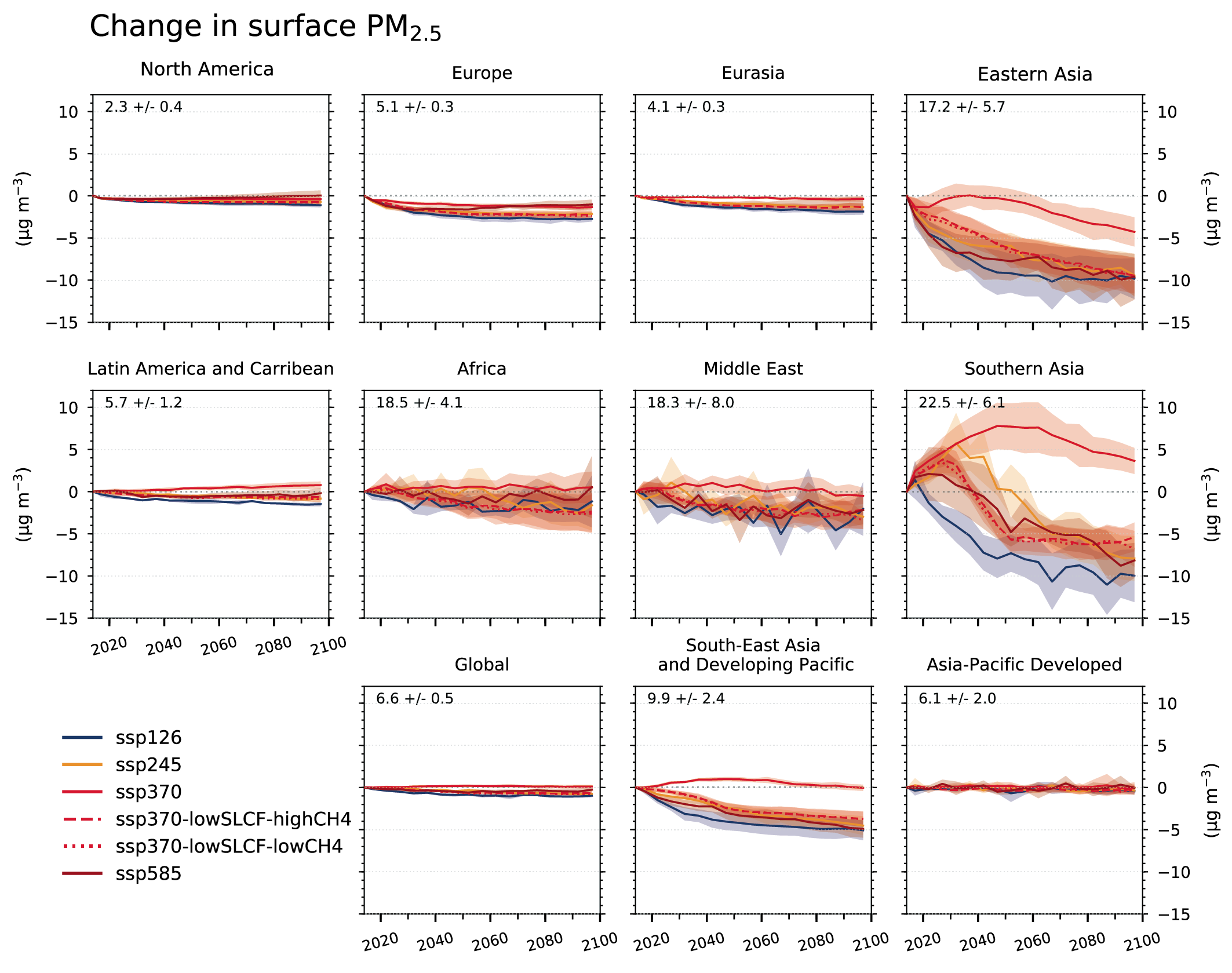 Figure 6.21 | Future changes in regional five-year mean surface PM2.5from 2015 to 2100 in different shared socio-economic pathways (SSPs). PM2.5 stands for micrograms per cubic meter of aerosols with diameter less than 2.5 μm and is calculated by summing up individual aerosol mass components from each model as: black carbon + particulate organic matter + sulphate + 0.25 × sea salt + 0.1 × dust. Since not all CMIP6 models reported nitrate aerosol, it is not included here. See Figure 6.20 for further details. PM2.5 changes are also displayed in the Interactive Atlas. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.21 | Future changes in regional five-year mean surface PM2.5from 2015 to 2100 in different shared socio-economic pathways (SSPs). PM2.5 stands for micrograms per cubic meter of aerosols with diameter less than 2.5 μm and is calculated by summing up individual aerosol mass components from each model as: black carbon + particulate organic matter + sulphate + 0.25 × sea salt + 0.1 × dust. Since not all CMIP6 models reported nitrate aerosol, it is not included here. See Figure 6.20 for further details. PM2.5 changes are also displayed in the Interactive Atlas. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). The magnitude of the annual mean change in surface ozone and PM2.5 for all the SSPs (accounting for both emissions and climate change) is greater than that expected from climate change in isolation (Turnock et al., 2020). The uncertainty in the projections comes from how natural emissions will respond to climate change. However, multiple lines of evidence (along with Sections 6.2.2, 6.5, and 6.7.1) provide high confidence (compared to medium in AR5) that changes in emissions, and in particular in human-induced emissions, will drive future air pollution levels rather than physical climate change.
In summary, future air pollution levels are strongly driven by precursor emissions trajectories in the SSPs with substantial reductions in global surface ozone and PM (when air pollution and climate change are both strongly mitigated, e.g., SSP1-2.6) to no improvement and even degradation (high confidence) (when no climate change mitigation and only weak air pollution control are considered, SSP3-7.0). In the latter case, PM levels are estimated to increase until 2050 over large parts of Asia and surface ozone pollution worsens over all continental areas throughout the whole century (high confidence). In scenarios without climate change mitigation but with strong air pollution control (SSP5-8.5), high methane levels hamper the decline in global surface ozone in the near term and only PM levels decrease (high confidence).
6.7.2 Evolution of Future Climate in Response to Changes in SLCF Emissions
6.7.2.1 Effects of changes in SLCFs on ERF and Climate Response
This section assesses how the different spatial and temporal evolution of SLCF emissions in the SSPs affects the future global and regional ERFs, and GSAT and precipitation responses. In CMIP6, only a very limited set of simulations (all based on the SSP3-7.0 scenario) have been carried out with coupled ESMs to specifically address the future role of SLCFs (Sections 4.3 and 4.4; Collins et al., 2017). Note that the ScenarioMIP simulations (Section 4.3) include the SLCF emissions (as shown in Figures 6.18 and 6.19), however, they cannot be used to quantify the effect of individual forcers. Coupled ESMs can in principle be used for this through a series of sensitivity simulations (e.g., Allen et al., 2020, 2021), but the amount of computer time required has made this approach prohibitive across the full SSP range. Therefore, to quantify the contribution from emissions of individual forcers spanning the range of the SSP scenarios to GSAT response, the analysis is mainly based on estimates using a two-layer emulator configuration derived from the medians of MAGICC7 and FaIRv1.6.2 (Section 1.5.3.4, Cross-Chapter Box 7.1 and Supplementary Material 7.SM5.2). The contribution from SLCFs to changes in GSAT have been calculated based on the global mean ERF for the various components as assessed in Section 7.3.5, using the two-layer emulator for the climate response.
The projections of GSAT for a broad group of forcing agents (aerosols, methane, tropospheric ozone and HFCs with lifetimes lower than 50 years) for the SSP scenarios show how much of the future warming or cooling (relative to 2019) can be attributed to the SLCFs (Figure 6.22). Note that during the first two decades, some of these changes in GSAT are due to emissions before 2019, in particular for the longer-lived SLCFs such as methane and HFCs (Figure 6.15). The scenarios SSP3-7.0-lowSLCF-highCH4 and SSP3-7.0-lowSLCF-lowCH4 are special cases of the SSP3-7.0 scenario with strong, but realistic, reductions in non-methane SLCFs and all SLCFs, respectively (Gidden et al., 2019).
As discussed in Sections 6.2, 6.3 and 6.4, there are uncertainties relating emissions of SLCFs to changes in abundance (Box 6.2) and further to ERF, in particular for aerosols and tropospheric ozone. Furthermore, there are uncertainties related to climate sensitivity, that is, the relation between ERF and change in GSAT. Uncertainties in the ERF are assessed in Chapter 7 and calibrated impulse response function also includes the assessed range (Box 7.1). There are also uncertainties related to the radiative efficacies of the different SLCFs and time scales for the response, in particular for regional emissions (Schwarber et al., 2019; Yang et al., 2019b) that cannot be accounted for with the simple models used here.
Historical emissions have been updated until 2019 (see Supplementary Material 7.SM.1.3.1) and used for ERF for calculating GSAT in Figure 6.22. The year 2019 has been chosen as the base year to be consistent with the attributed temperature changes since 1750 (Figure 7.8). The warming attributed to SLCFs (methane, ozone and aerosols) over the last decade (Figure 7.8) constitutes about 30% of the peak SLCF-driven warming in the most stringent scenarios (SSP1), in good agreement with Shindell and Smith (2019), and supported by the recent observed decline in AOD (Section 2.2.6).
From 2019 and until about 2040, SLCFs and HFCs will contribute to increase GSAT in the WGI core set of SSP scenarios, with a very likely range of 0.04°C–0.41°C relative to 2019. The warming is most pronounced in the strong mitigation scenarios (i.e., SSP1-1.9 and SSP1-2.6) due to rapid cuts in aerosols. In scenario SSP3-7.0, there is no reduction of aerosols until mid-century and it is the increases in methane and ozone that give a net warming in 2040. The warming is similar in magnitude to that in the SSP1-scenarios, in which the reduction in aerosols is the main driver. Contributions to warming from methane, ozone, aerosols and HFCs make SSP5-8.5 the scenario with the highest warming in 2040 and throughout the century.
After about 2040, it is likely that, across the scenarios, the net effect of the removal of aerosols is a further increase in GSAT. However, their contribution to the rate of change decreases towards the end of the century (from up to 0.2°C per decade before 2040 to about 0.03°C per decade after 2040). After 2040, the changes in methane, HFCs and tropospheric ozone become equally important as the changes in the aerosols for the GSAT trends. In the low-emissions scenarios (SSP1-1.9 and SSP1-2.6), the contribution to warming from the SLCFs peaks around 2040 with a very likely range of 0.04°C to 0.34°C. After the peak, the reduced warming from reductions in methane and ozone dominates, giving a best total estimate warming induced by SLCF and HFC changes of 0.12°C and 0.14°C respectively, in 2100, with a very likely range of –0.07°C to +0.45°C (Figure 6.22). However, in the longer term towards the end of the century there are very significant differences between the scenarios. In SSP3-7.0 there is a near-linear warming due to SLCFs of 0.08°C per decade, while for SSP5-8.5 there is a more rapid early warming. In SSP3-7.0, the limited reductions in aerosols, but a steady increase in methane, HFCs and ozone lead to a nearly linear contribution to GSAT reaching a best estimate of 0.5°C in 2100. Contributions from methane and ozone decrease towards 2100 in SSP5-8.5, however the warming from HFCs still increase and the SSP5-8.5 has the largest SLCF and HFC warming in 2100 with a best estimate of 0.6°C. In the SSP2-4.5 scenario, a reduction in aerosols contributes to about 0.3°C warming in 2100, while contributions from ozone and methane in this scenario are small.
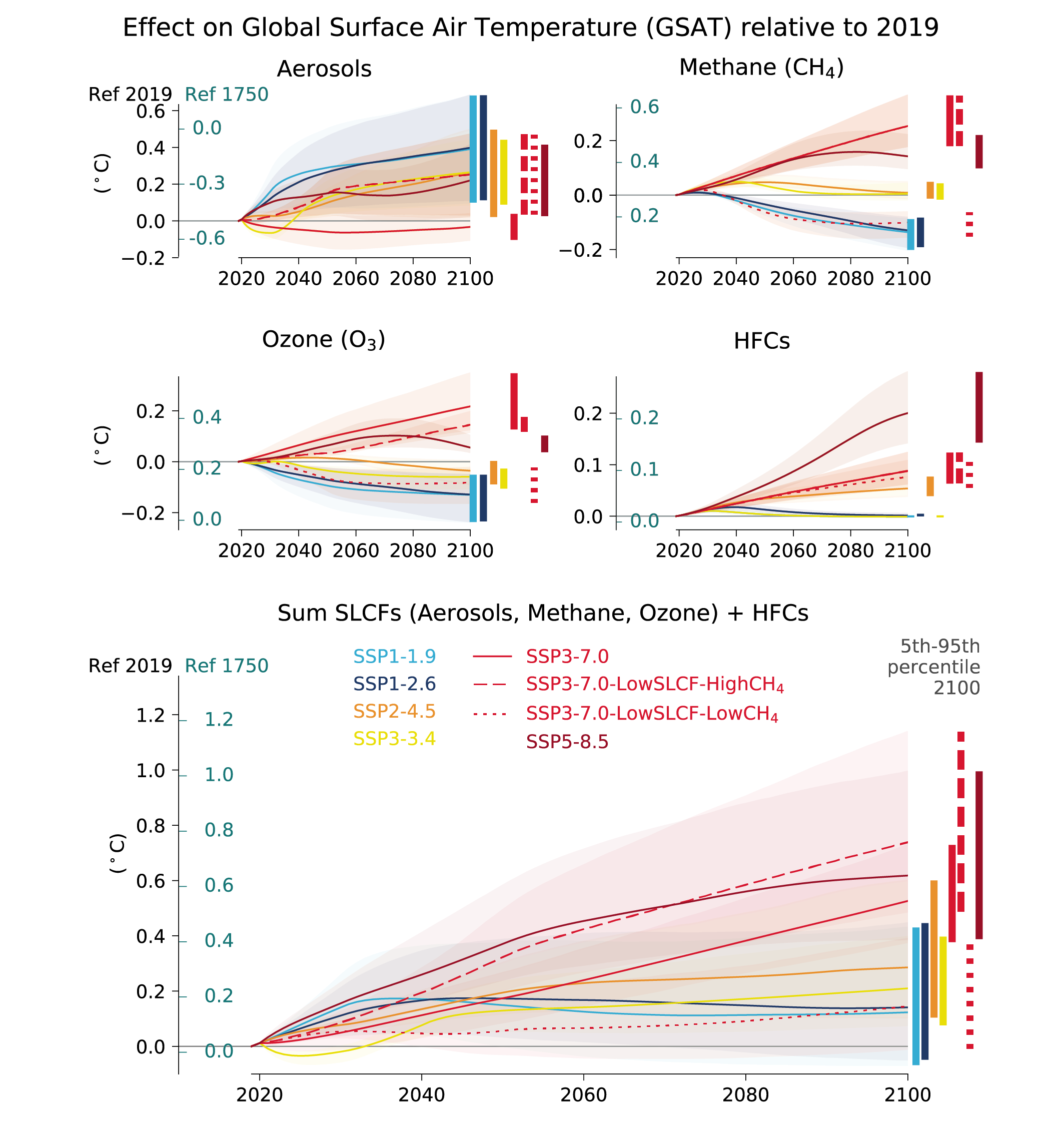 Figure 6.22 | Time evolution of the effects of changes in short-lived climate forcers (SLCFs) and hydrofluorocarbons (HFCs) on global surface air temperature (GSAT) across the WGI core set of Shared Socio-Economic Pathways (SSPs). Effects of net aerosols, methane, tropospheric ozone and hydrofluorocarbons (HFCs; with lifetimes <50years), and the sum of these, relative to the year 2019 and to the year 1750. The GSAT changes are based on the assessed historic and future evolution of effective radiative forcing (ERF; Section 7.3.5). The temperature responses to the ERFs are calculated with an impulse response function with an equilibrium climate sensitivity of 3.0°C for a doubling of atmospheric CO2 (feedback parameter of –1.31 W m–2°C–1, see Cross-Chapter Box 7.1). The vertical bars to the right in each panel show the uncertainties (5–95% ranges) for the GSAT change between 2019 and 2100. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.22 | Time evolution of the effects of changes in short-lived climate forcers (SLCFs) and hydrofluorocarbons (HFCs) on global surface air temperature (GSAT) across the WGI core set of Shared Socio-Economic Pathways (SSPs). Effects of net aerosols, methane, tropospheric ozone and hydrofluorocarbons (HFCs; with lifetimes <50years), and the sum of these, relative to the year 2019 and to the year 1750. The GSAT changes are based on the assessed historic and future evolution of effective radiative forcing (ERF; Section 7.3.5). The temperature responses to the ERFs are calculated with an impulse response function with an equilibrium climate sensitivity of 3.0°C for a doubling of atmospheric CO2 (feedback parameter of –1.31 W m–2°C–1, see Cross-Chapter Box 7.1). The vertical bars to the right in each panel show the uncertainties (5–95% ranges) for the GSAT change between 2019 and 2100. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). The simplified approach used to estimate the contributions to GSAT in Figure 6.22 has been supplemented with ESM simulations driven by the two versions of the SSP3-7.0-lowSLCF scenario (Section 6.7.1.1). Results from five CMIP6 ESMs with fully interactive atmospheric chemistry and aerosols for the high-methane scenario show (Allen et al., 2020, 2021) that reductions in emissions of air pollutants would lead to an additional increase in GSAT by 2055 relative to 2015 compared to the standard SSP3-7.0 scenario, with a best estimate of 0.23°C ± 0.05°C, and a corresponding increase in global mean precipitation of 1.3 ± 0.17% (note that uncertainties from the work of Allen et al. here and elsewhere are reported as twice standard deviation). Including methane mitigation (SSP3-7.0-lowSLCF-lowCH4) would lead to a small increase in global precipitation (0.7 ± 0.1%) by mid-century despite a decrease in GSAT (Section 6.7.3), which is related to the higher sensitivity of precipitation to sulphate aerosols than greenhouse gases (Section 8.2.1; Allen et al., 2021).
Regionally inhomogeneous ERFs can lead to regionally dependent responses (Section 6.4.3). Mitigation of non-methane SLCFs over the period 2015–2055 (SSP3-7.0-lowSLCF-highCH4 versus SSP3-7.0) will lead to positive ERF over land regions (Allen et al. , 2020). There are large regional differences in the ERF from no significant trend over northern Africa to about 0.5 W m–2 decade–1 for Southern Asia. The differences are mainly driven by differences in the reductions of sulphate aerosols. There is no strong correspondence between regional warming and the ERF trends. As expected, the sensitivity (temperature change per unit ERF) increases towards higher latitudes due to climate feedbacks and teleconnections. Regionally, the warming rates are higher over continental regions, with the highest increase in temperatures for Central and northern Asia and the Arctic in 2055 relative to 2015. The models agree on an increasing global mean trend in precipitation due to SLCFs, however precipitation trends over land are more uncertain (Allen et al., 2020), in agreement with the relationship between aerosol and precipitation trends assessed in Chapter 8.
ESM estimates of future concentrations of various SLCFs vary considerably even when using the same future emissions scenarios, which is related to sources of model structural uncertainty in the several physical, chemical and natural emissions model parametrizations. The general uncertainties in understanding and representing chemical and physical processes governing the life cycle of SLCFs (Box 6.1) necessarily also applies to simulations of future concentrations and ERF. In addition, how the models are able to simulate climate changes (i.e., circulation and precipitation) that affect the dispersion and removal of SLCFs constitute a structural uncertainty in the models. Also SLCF-related climate feedbacks (e.g., NOx from lightning or BVOCs from vegetation; Section 6.4.5) add to the uncertainty.
In the near term (2035–2040), it is unlikely that differences in the socio-economic developments and emissions controls induced by policies (as embedded in the SSPs) can lead to a discernible difference in the net effect of changes of SLCFs on GSAT. This is because the inter-model spread in the estimated net effect of SLCFs on GSAT is as large as the difference between the scenarios due to the compensating effects of change in emissions leading to cooling and warming. However, in the longer term, there is high confidence that the net warming induced by changes in SLCFs will be lower in the scenario considering strong climate change mitigation (SSP1-1.9 and SSP1-2.6 that include reductions in methane emissions) than in the high CO2 emissions scenarios (SSP3-7.0 and SSP5-8.5).
6.7.2.2 Effect of Regional Emissions of SLCFs on GSAT
For SLCFs with lifetimes shorter than typical mixing times in the atmosphere (days to weeks), the effects on secondary forcing agents (e.g., tropospheric ozone, sulphate and nitrate aerosols) depend on where and when the emissions occur due to non-linear chemical and physical processes. Also, the ERF following a change in concentrations depends on the local conditions (Sections 6.2, 6.3 and 6.4).
While the emulators used for GSAT projections shown in Figure 6.22 do not take the regional perspective into account, the set of simulations performed within the Hemispheric Transport of Air Pollutants Phase 2 (HTAP2) project (Galmarini et al., 2017) allows for this perspective. The results from the chemistry–transport model OsloCTM3 taking part in the HTAP2 have been used by Lund et al. (2020) to derive region-specific absolute global warming potentials (AGTPs; cf. Aamaas et al., 2016) for each emitted SLCF and each HTAP2 region. With this set of AGTPs, Lund et al. (2020) estimate the transient response in GSAT to the regional anthropogenic emissions. There are important differences in the contributions to GSAT in 2040 and 2100 (relative to 2020) between the regions and scenarios, mainly due to the differences in the mixture of emitted SLCFs (Figure 6.23). There is overall good agreement between the total net contribution from all regions to GSAT and the estimate based on global ERF and the two-layer emulator (Figure 6.22).
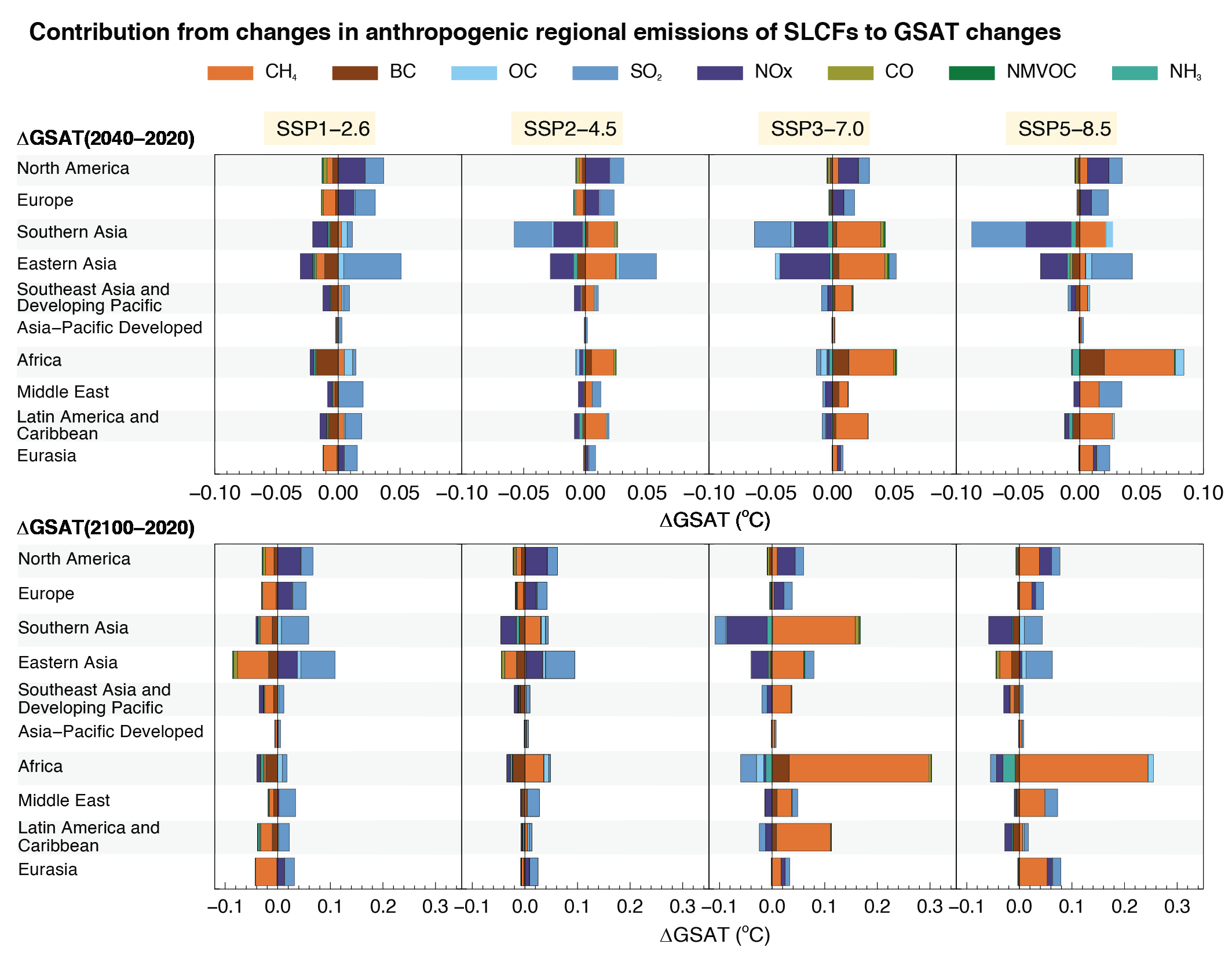 Figure 6.23 | Contribution from regional emissions of short-lived climate forcers (SLCFs) to changes in global surface air temperature (GSAT) in 2040 (upper row) and 2100 (lower row), relative to 2020 for four Shared Socio-economic Pathways (SSPs). Adapted from Lund et al. (2020). NOx, CO, and NMVOC account for the impact through changes in ozone and methane, NOx additionally includes the impact through formation of nitrate aerosols. BC, SO2 and OC accounts for the direct aerosol effect (aerosol–radiation interactions), as well as an estimate of the semi-direct effect for BC due to rapid adjustments and indirect effect (aerosol–cloud interactions) of sulphate aerosols. Regions are the same as shown in the map in Figure 6.7. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.23 | Contribution from regional emissions of short-lived climate forcers (SLCFs) to changes in global surface air temperature (GSAT) in 2040 (upper row) and 2100 (lower row), relative to 2020 for four Shared Socio-economic Pathways (SSPs). Adapted from Lund et al. (2020). NOx, CO, and NMVOC account for the impact through changes in ozone and methane, NOx additionally includes the impact through formation of nitrate aerosols. BC, SO2 and OC accounts for the direct aerosol effect (aerosol–radiation interactions), as well as an estimate of the semi-direct effect for BC due to rapid adjustments and indirect effect (aerosol–cloud interactions) of sulphate aerosols. Regions are the same as shown in the map in Figure 6.7. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). In the low- and medium-emissions scenarios (SSP1-2.6 and SSP2-4.5), the warming effects induced by changes in SLCFs on GSAT are dominated by emissions in North America, Europe and Eastern Asia (Figure 6.23). In SSP1-2.6 the emissions of all SLCFs in all regions decrease and the net effect of the changes in SLCFs from all of these three regions is an increase in GSAT of about 0.02°C (per region) in 2040 and about 0.04°C in 2100. For SSP2-4.5, emissions of most SLCFs continue to increase in Southern Asia (Figure 6.19), leading to a net cooling in the near term (–0.03°C in 2040), while in 2100, North America, Europe, Eastern and Southern Asia all contribute to a warming, most pronounced from Eastern Asia (0.05°C). In the SSP3-7.0 scenario, the net effect induced by changes in SLCFsin allregions is an enhanced warming towards the end of the century, drivenpredominantly by change in methane. Africa is the region contributing the most topredictedglobal warmingdue to SLCF changesin2100(0.24°C). In SSP5-8.5, methane emissions increase in North America, Europe and Africa, while there is a decrease in the Asian regions. For North America and Europe, the methane increase combined with a reduction in aerosol leads to highest net contribution to GSAT in this scenario (0.06°C and 0.04°C in 2100, respectively). The high growth in methane makes Africa the region with the largest contribution to future warming by SLCFs (0.18°C in 2100 versus 2020) in this scenario.
6.7.3 Effect of SLCF Mitigation in SSP Scenarios
Air-quality policies lead to a decrease in emissions of both warming and cooling SLCFs. Here we assess the contribution of SLCFs to the total warming (also including the LLGHGs) in the case of stringent SLCF mitigation to improve air quality in scenarios with continued high use of fossil fuels (e.g., SSP3-7.0-lowSLCF and SSP5-8.5). Conversely, we also assess the effect on air quality of strategies aiming to mitigate air pollution or climate change under the SSP3-7.0 framework (using the SSP3-7.0-lowSLCF-lowCH4, SSP3-7.0-lowSLCF-highCH4 and SSP3-3.4 scenarios).
As illustrated in Figure 2.2 of SR1.5 (Rogelj et al., 2018a), the total aerosol ERF change in stringent mitigation pathways is expected to be positive and to contribute to a warming since it is dominated by the effects from the phase-out of SO 2 (Figure 6.24, Section 6.7.2.2). Recent emissions inventories and observations of trends in AOD (Sections 2.2.6 and 6.2.1) show that it is very likely that there have been reductions in global SO2 emissions and in aerosol burdens over the last decade. Here, we use 2019 as the reference year rather than the ‘Recent Past’ defined as the average over 1995–2014 (Section 4.1) in order to exclude the recent emissions reductions when discussing the different possible futures.
The role of the different SLCFs, and also the net of all the SLCFs relative to the total warming in the scenarios, is quite different across the SSP scenarios varying with the summed levels of climate change mitigation and air pollution control (Figure 6.24). In the scenario without climate change mitigation but with strong air pollution control (SSP5-8.5) all the SLCFs (methane, aerosols and tropospheric ozone) and the HFCs (with lifetimes less than 50 years) add to the warming, while in the strong climate change and air pollution mitigation scenarios (SSP1-1.9 and SSP1-2.6), the emissions controls act to reduce methane, ozone and BC, and these reductions thus contribute to cooling. In all scenarios, except SSP3-7.0, emissions controls lead to a reduction of the aerosols relative to 2019, causing a warming. However, the warming from aerosol reductions is stronger in the SSP1 scenarios (with best estimates of 0.21°C in 2040 and 0.4°C in 2100 in SSP1-2.6) because of higher emissions reductions from stronger decrease of fossil fuel use in these scenarios than in SSP5-8.5 (0.13°C in 2040 and 0.22°C in 2100). The changes in methane abundance contribute a warming of 0.14°C in SSP5-8.5, but a cooling of 0.14°C in SSP1-2.6 by the end of the 21st century relative to 2019. Furthermore, under SSP5-8.5, HFCs induce a warming of 0.06°C with a very likely range of [0.04 to 0.08] °C in 2050 and 0.2 [0.1 to 0.3] °C by the end of the 21st century, relative to 2019, while under SSP1-2.6, warming due to HFCs is negligible (below 0.02°C) (high confidence). This assessment relies on these estimates, which are based on updated ERFs and HFC lifetimes. It is in accordance with previous estimates (Section 6.6.3.2) of the efficiency of the implementation of the Kigali Amendment and national regulations. It is very likely that under a stringent climate and air pollution mitigation scenario (SSP1-2.6), the warming induced by changes in methane, ozone, aerosols and HFCs towards the end of the 21st century, will be very low compared with the warming they would cause under the SSP5-8.5 scenario (0.14°C in SSP1-2.6 versus 0.62°C in SSP5-8.5).
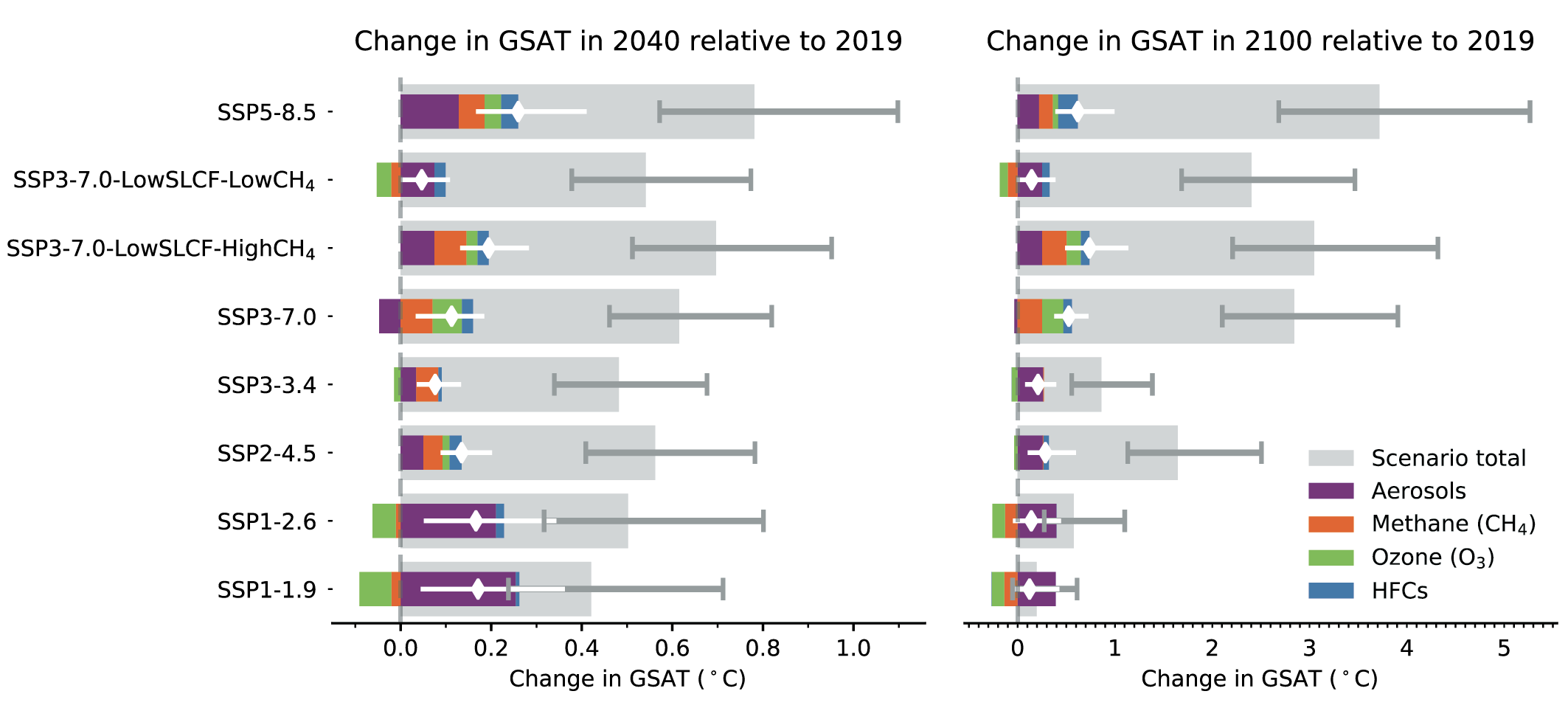 Figure 6.24 | Effects of changes in short-lived climate forcers (SLCFs) and hydrofluorocarbons (HFCs) on global surface air temperature (GSAT) across the WGI core set of Shared Socio-economic Pathways (SSPs). Effects of net aerosols, methane, tropospheric ozone and hydrofluorocarbons (HFCs; with lifetimes <50years), are compared with those of total anthropogenic forcing for 2040 and 2100 relative to the year 2019. The GSAT changes are based on the assessed historic and future evolution of effective radiative forcing (ERF; Section 7.3.5). The temperature responses to the ERFs are calculated with an impulse response function with an equilibrium climate sensitivity of 3.0°C for a doubling of atmospheric CO2 (feedback parameter of –1.31 W m–2°C–1; Cross-Chapter Box 7.1). Uncertainties are 5–95% ranges. The scenario total (grey bar) includes all anthropogenic forcings (long- and short-lived climate forcers, and land-use changes) whereas the white diamonds and bars show the net effects of SLCFs and HFCs and their uncertainties. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.24 | Effects of changes in short-lived climate forcers (SLCFs) and hydrofluorocarbons (HFCs) on global surface air temperature (GSAT) across the WGI core set of Shared Socio-economic Pathways (SSPs). Effects of net aerosols, methane, tropospheric ozone and hydrofluorocarbons (HFCs; with lifetimes <50years), are compared with those of total anthropogenic forcing for 2040 and 2100 relative to the year 2019. The GSAT changes are based on the assessed historic and future evolution of effective radiative forcing (ERF; Section 7.3.5). The temperature responses to the ERFs are calculated with an impulse response function with an equilibrium climate sensitivity of 3.0°C for a doubling of atmospheric CO2 (feedback parameter of –1.31 W m–2°C–1; Cross-Chapter Box 7.1). Uncertainties are 5–95% ranges. The scenario total (grey bar) includes all anthropogenic forcings (long- and short-lived climate forcers, and land-use changes) whereas the white diamonds and bars show the net effects of SLCFs and HFCs and their uncertainties. Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). For the SSP3-7.0-lowSLCF-highCH4 and SSP3-7.0-lowSLCF-lowCH4 scenarios, a five-ESM ensemble has been analysed relative to the standard SSP3-7.0 scenario (Allen et al., 2020, 2021). For SSP3-7.0-lowSLCF-highCH4, in which the methane emissions are as in the standard SSP3-7.0 scenario, Allen et al. (2021) found an enhanced global mean surface warming of 0.23°C ± 0.05°C by mid-century and 0.21°C ± 0.03°C by 2100 relative to the warming in the standard SSP3-7.0 scenario. Also including strong mitigation of methane emissions, the same models (Allen et al., 2021) find that the warming is offset resulting in a net cooling of 0.15°C ± 0.05°C at mid-century (2050–2059) and 0.50°C ± 0.02°C at the end of the century (2090–2099) relative to SSP3-7.0.
There is robust evidence and high agreement that non-methane SLCF mitigation measures, through reductions in aerosols and non-methane ozone precursors to improve air quality (SSP3-7.0-lowSLCF-highCH4 vs SSP3-7.0), would lead to additional near-term warming with a range of 0.1°C–0.3°C. Methane mitigation that also reduces tropospheric ozone, stands out as an option that combines near- and long-term gains on surface temperature (high confidence). With stringent but realistic methane mitigation (SSP3-7.0-lowSLCF-lowCH4), it is very likely that warming (relative to SSP3-7.0) from non-methane SLCFs can be offset (Figure 6.24; Allen et al., 2021). Due to the slower response to the methane mitigation, this offset becomes more robust over time and it is very likely to be an offset after 2050. However, when comparing to the present day, it is unlikely that methane mitigation can fully cancel out the warming over the 21st century from reduction of non-methane cooling SLCFs.
The SSP3 storyline assumes ‘regional rivalry’ (Section 1.6.1.1) with weak air pollution legislation and no climate change mitigation, and is compared here against SSP3-7.0-lowSLCF-lowCH4 (strong air pollution control) and SSP3-3.4 (the most ambitious climate policy feasible under the SSP3 narrative). In the SSP3-3.4 scenario, all emissions follow the SSP3-7.0 scenario until about 2030 and then deep and rapid cuts in fossil fuel use are imposed (Fujimori et al., 2017). In the case of climate change mitigation, such as in the SSP3-3.4 scenario, the decrease of SLCF emissions is a co-benefit from the targeted decrease of CO2 (when SLCFs are co-emitted), but also directly targeted as in the case of methane. For SLCFs, this means that emissions of aerosols and methane increase until 2030 and are reduced quickly thereafter (Fujimori et al., 2017). The effect on GSAT (relative to 2019) is shown in Figures 6.22 and 6.24. The net GSAT response to the SLCFs is dominated by the aerosols, with an initial cooling until 2030, then a fast rebound for 15 years followed by a very moderate warming reaching 0.21°C in 2100. The ozone reduction causes a slight cooling (up to 0.06°C), in contrast to the warming in the SSP3-7.0-lowSLCF-highCH4 scenario in which the methane emissions increase until 2100.
To assess the effect of dedicated air-quality versus climate policy on air quality, PM2.5 and ozone indicators were estimated for three SSP3 scenarios by applying a widely used approach for the analysis of air-quality implications for given emissions scenarios (Rao et al., 2017; Van Dingenen et al., 2018; Vandyck et al., 2018) and whose sensitivity of surface concentrations to emissions changes is comparable to that in the ESM ensemble (Supplementary Material 6.SM.5). The assessment shows that both strong air pollution control and strong climate change mitigation, implemented independently, lead to a large reduction of exposure to PM2.5 and ozone by the end of the century (high confidence) (Figures 6.25 and 6.26). However, implementation of air pollution control, relying on the deployment of existing technologies, leads to benefits more rapidly than climate change mitigation (high confidence), which requires systemic changes and is thus implemented later in this scenario. Notably, under the underlying SSP3 context, significant parts of the population remain exposed to air quality exceeding the WHO guidelines for PM2.5 over the whole century (high confidence), in particular in Africa, Eastern and Southern Asia and the Middle East, and for ozone only a small improvement in population exposure is expected in Africa and Asia. Confidence levels here result from expert judgement on the whole chain of evidence.
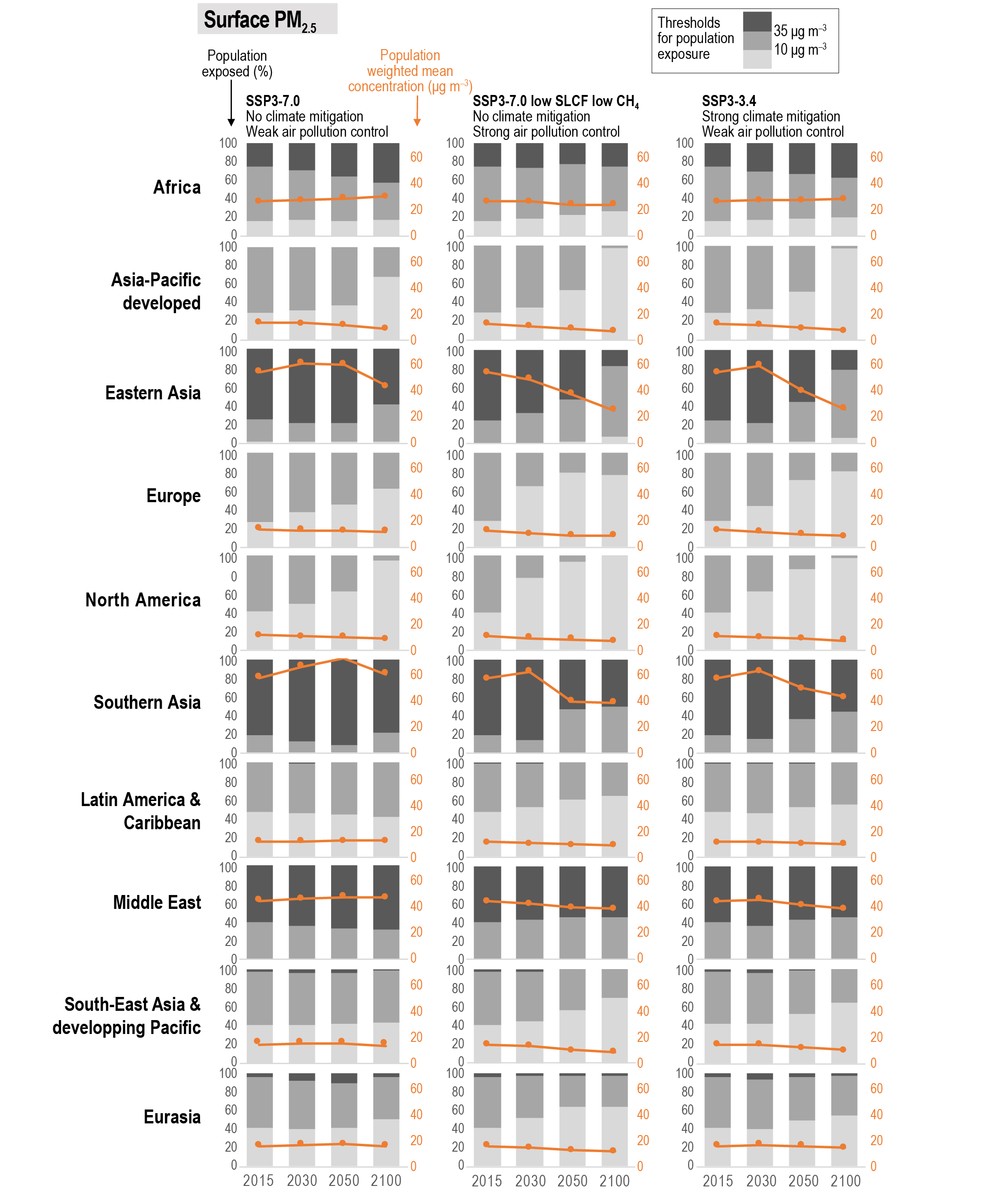 Figure 6.25 | Effect of dedicated air pollution or climate policy on population-weighted PM2.5 concentrations (µg m–3) and share of population (%) exposed to different PM2.5levels across 10 world regions. Thresholds of 10 µg m–3 and 35 µg m–3represent the WHO air quality guideline and the WHO interim target 1, respectively; WHO (2017). Results are compared for SSP3-7.0 (no major improvement of current legislation is assumed), SSP3-lowSLCF (strong air pollution controls are assumed), and a climate change mitigation scenario SSP3-3.4; details of scenario assumptions are discussed in Riahi et al. (2017) and Rao et al. (2017). Analysis performed with the TM5-FASST model (Van Dingenen et al., 2018) using emissions projections from the Shared economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.25 | Effect of dedicated air pollution or climate policy on population-weighted PM2.5 concentrations (µg m–3) and share of population (%) exposed to different PM2.5levels across 10 world regions. Thresholds of 10 µg m–3 and 35 µg m–3represent the WHO air quality guideline and the WHO interim target 1, respectively; WHO (2017). Results are compared for SSP3-7.0 (no major improvement of current legislation is assumed), SSP3-lowSLCF (strong air pollution controls are assumed), and a climate change mitigation scenario SSP3-3.4; details of scenario assumptions are discussed in Riahi et al. (2017) and Rao et al. (2017). Analysis performed with the TM5-FASST model (Van Dingenen et al., 2018) using emissions projections from the Shared economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). 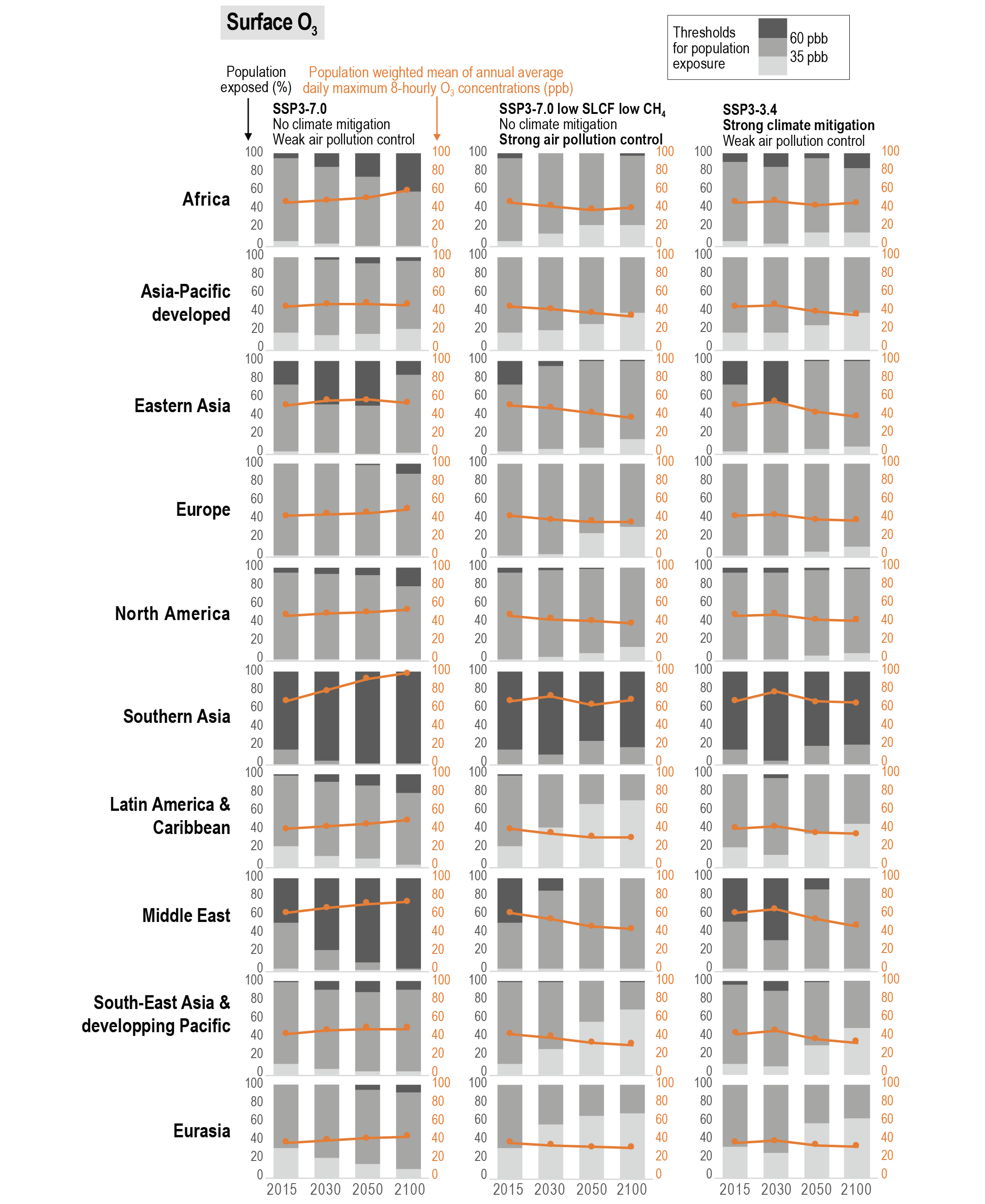 Figure 6.26 | Effect of dedicated air pollution or climate policy on population-weighted ozone concentrations (ppb) and share of population (%) exposed to different ozone levels across 10 world regions. Results are compared for SSP3-7.0 (no major improvement of current legislation is assumed), SSP3-low SLCF (strong air pollution controls are assumed), and a climate change mitigation scenario (SSP3-3.4); details of scenario assumptions are discussed in Riahi et al. (2017) and Rao et al. (2017). Analysis performed with the TM5-FASST model (Van Dingenen et al., 2018) using emissions projections from the Socio-economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3).
Figure 6.26 | Effect of dedicated air pollution or climate policy on population-weighted ozone concentrations (ppb) and share of population (%) exposed to different ozone levels across 10 world regions. Results are compared for SSP3-7.0 (no major improvement of current legislation is assumed), SSP3-low SLCF (strong air pollution controls are assumed), and a climate change mitigation scenario (SSP3-3.4); details of scenario assumptions are discussed in Riahi et al. (2017) and Rao et al. (2017). Analysis performed with the TM5-FASST model (Van Dingenen et al., 2018) using emissions projections from the Socio-economic Pathway (SSP) database (Riahi et al., 2017; Rogelj et al., 2018a; Gidden et al., 2019). Further details on data sources and processing are available in the chapter data table (Table 6.SM.3). In summary, the warming induced by SLCF changes is stable after 2040 in the WGI core set of SSP scenarios associated with lower global air pollution as long as methane emissions are also mitigated, but the overall warming induced by SLCF changes is higher in scenarios in which air quality continues to deteriorate (caused by growing fossil fuel use and limited air pollution control) (high confidence). In the SSP3-7.0 context, applying an additional strong air pollution control resulting in reductions in anthropogenic aerosols and non-methane ozone precursors would lead to an additional near-term global warming of 0.08°C with a very likely range of [–0.05 to 0.25] °C (compared with SSP3-7.0 for the same period). A simultaneous methane mitigation consistent with SSP1’s stringent climate change mitigation policy implemented in the SSP3 world, could entirely alleviate this warming and even lead to a cooling of 0.07°C with a very likely range of [–0.08 to +0.18] °C (compared withSSP3-7.0 for the same period). Across the SSPs, the reduction of methane, ozone precursors and HFCs can make a 0.2 [0.1 to 0.4] °C difference on GSAT in 2040 and a 0.8 [0.5 to 1.3] °C difference at the end of the 21st century (Figure 6.24), which is substantial in the context of the Paris Agreement. Sustained methane mitigation, wherever it occurs, stands out as an option that combines near- and long-term gains on surface temperature (high confidence) and leads to air pollution benefits by reducing surface ozone level globally (high confidence).
Strong air pollution control as well as strong climate change mitigation, implemented independently, lead to large reduction of the exposure to air pollution by the end of the century (high confidence). Implementation of air pollution control, relying on the deployment of existing technologies, leads more rapidly to air-quality benefits than climate change mitigation which requires systemic changes but, in both cases, significant parts of the population remain exposed to air pollution exceeding the WHO guidelines (high confidence).
6.8 Perspectives
Ice-core analyses can now inform trends for more SLCFs over the last millennium (such as light NMVOCs or CO) and more proxies are available to inform about past emissions. However, pre-industrial levels of SLCFs are still relatively poorly constrained. In addition, recent trends in abundances of the various types of aerosols and of NMVOCs suffer from the scarcity of observation networks in various parts of the world, in particular in the Southern Hemisphere. Such network development is necessary to record and understand the evolution of atmospheric composition.
Assessment of future air pollution changes at the urban level requires the use of a high-resolution models to properly account for non-linearities in chemistry, specific urban structures and local meteorology as well as temporal and spatial variations in emissions and population exposure. To assess the relevance of air pollution reduction policies, regional air-quality models are necessary and are still not implemented in many developing countries. To properly apply such models, the quality of spatialized emissions inventories is essential, but the production of accurate emissions inventories remain a challenge for lots of rapidly growing urban areas. The emissions reporting now planned in the official mandate of the Task Force on National Greenhouse Gas Inventories (TFI) can be a step in this direction if accompanied by efforts on spatial distribution of emissions. An integrated modelling framework associating global and high-resolution chemistry–transport models with shared protocols is missing to allow a systematic assessment of future changes on air quality at this scale.
In parallel, opportunities of progress may emerge from big-data acquisition. Big data and their mining can inform practices related to emissions or can document pollution levels if associated with massive deployment of low-cost sensors through citizen science. New generation satellite data will also give access to sub-kilometre-scale air pollution observations.
A systematic emissions modeling framework is needed to assess the LLGHG emissions changes associated with SLCF reductions induced by air pollution control in the SSP framework. The SLCF-mediated effects of large-scale technology deployment to allow climate change mitigation, such as hydrogen energy production, carbon capture and storage though amine filters, or changes in agricultural practices to limit GHG emissions and/or produce bioenergy are also not considered in the emissions scenarios.
Since AR5, the complexity of ESMs has increased to include many chemical and biogeochemical processes. These processes are necessary to quantify non-CO2 biogeochemical feedbacks on the Earth system resulting from climate-driven changes in atmospheric chemistry and SLCF emissions from natural systems and, in turn, the impact of SLCFs on biogeochemical cycles. Enhanced understanding of the biological, chemical and physical processes based on experimental and observational work has facilitated advances in the ESMs. However, assessment of non-CO2 biogeochemical feedbacks and SLCF effects on land and aquatic ecosystem productivity still remains challenging due to the multiple complex processes involved and limitations in observational constraints to evaluate the skill of ESMs in realistically simulating the processes. Advances will come from better understanding of the processes and mechanisms, in particular at the interfaces between components. The development of high-resolution ESMs will facilitate their evaluation against high- resolution observations.
Frequently Asked Questions
FAQ 6.1 | What Are Short-lived Climate Forcers and How Do They Affect the Climate?
Short-lived climate forcers (SLCFs) are compounds such as methane and sulphate aerosols that warm or cool the Earth’s climate over shorter time scales – from days to years – than greenhouse gases like carbon dioxide, whose climatic effect lasts for decades, centuries or more. Because SLCFs do not remain in the atmosphere for very long, their effects on the climate are different from one region to another and can change rapidly in response to changes in SLCF emissions. As some SLCFs also negatively affect air quality, measures to improve air quality have resulted in sharp reductions in emissions and concentrations of those SLCFs in many regions over the few last decades.
The SLCFs include gases as well as tiny particles called aerosols, and they can have a warming or cooling effect on the climate (FAQ 6.1, Figure 1). Warming SLCFs are either greenhouse gases (e.g., ozone or methane) or particles like black carbon (also known as soot), which warm the climate by absorbing energy and are sometimes referred to as short-lived climate pollutants. Cooling SLCFs, on the other hand, are mostly made of aerosol particles (e.g., sulphate, nitrate and organic aerosols) that cool down the climate by reflecting away more incoming sunlight.
Some SLCFs do not directly affect the climate but produce climate-active compounds and are referred to as precursors. SLCFs are emitted both naturally and as a result of human activities, such as agriculture or extraction of fossil fuels. Many of the human sources, particularly those involving combustion, produce SLCFs at the same time as carbon dioxide and other long-lived greenhouse gases. Emissions have increased since the start of industrialization, and humans are now the dominant source for several SLCFs and SLCF precursors, such as sulphur dioxide (which produces sulphate aerosols) and nitrogen oxides (which produce nitrate aerosols and ozone), despite strong reductions over the last few decades in some regions due to efforts to improve air quality.
The climatic effect of a chemical compound in the atmosphere depends on two things: (i) how effective it is at cooling or warming the climate (its radiative efficiency) and (ii) how long it remains in the atmosphere (its lifetime). Because they have high radiative efficiencies, SLCFs can have a strong effect on the climate even though they have relatively short lifetimes of up to about two decades after emission. Today, there is a balance between warming and cooling from SLCFs, but this can change in the future.
The short lifetime of SLCFs constrains their effects in both space and time. First, of all the SLCFs, methane and the short-lived halocarbons persist the longest in the atmosphere: up to two decades (FAQ 6.1, Figure 1). This is long enough to mix in the atmosphere and to spread globally. Most other SLCFs only remain in the atmosphere for a few days to weeks, which is generally too short for mixing in the atmosphere, sometimes even regionally. As a result, the SLCFs are unevenly distributed and their effects on the climate are more regional than those of longer-lived gases. Second, rapid (but sustained) changes in emissions of SLCFs can result in rapid climatic effects.
In addition to the direct warming and cooling effects, SLCFs have many other consequences for the climate system and for air quality (see FAQ 6.2). For instance, deposition of black carbon on snow darkens its surface, which subsequently absorbs more solar energy, leading to more melting and more warming. Aerosols also modify the properties of clouds, which has indirect cooling effects on the climate and causes changes in local rainfall (see FAQ 7.2). Climate models indicate that SLCFs have altered atmospheric circulation on local and even hemispheric scales (e.g., monsoons) as well as regional precipitation. For instance, recent observations show that regional weather is influenced by strong regional contrasts in the evolution of aerosol concentrations, particularly over South and East Asia.
Although policies to limit climate change and discussions of the so-called remaining carbon budgets primarily focus on carbon dioxide (see FAQ 5.4), SLCFs can significantly affect temperature changes. It is therefore important to understand how SLCFs work and to quantify their effects. Because reducing some of the SLCF emissions, such as methane, can simultaneously reduce warming effects and adverse effects on air quality as well as help attaining Sustainable Development Goals, mitigation of SLCFs is often viewed as a favourable ‘win-win’ policy option.
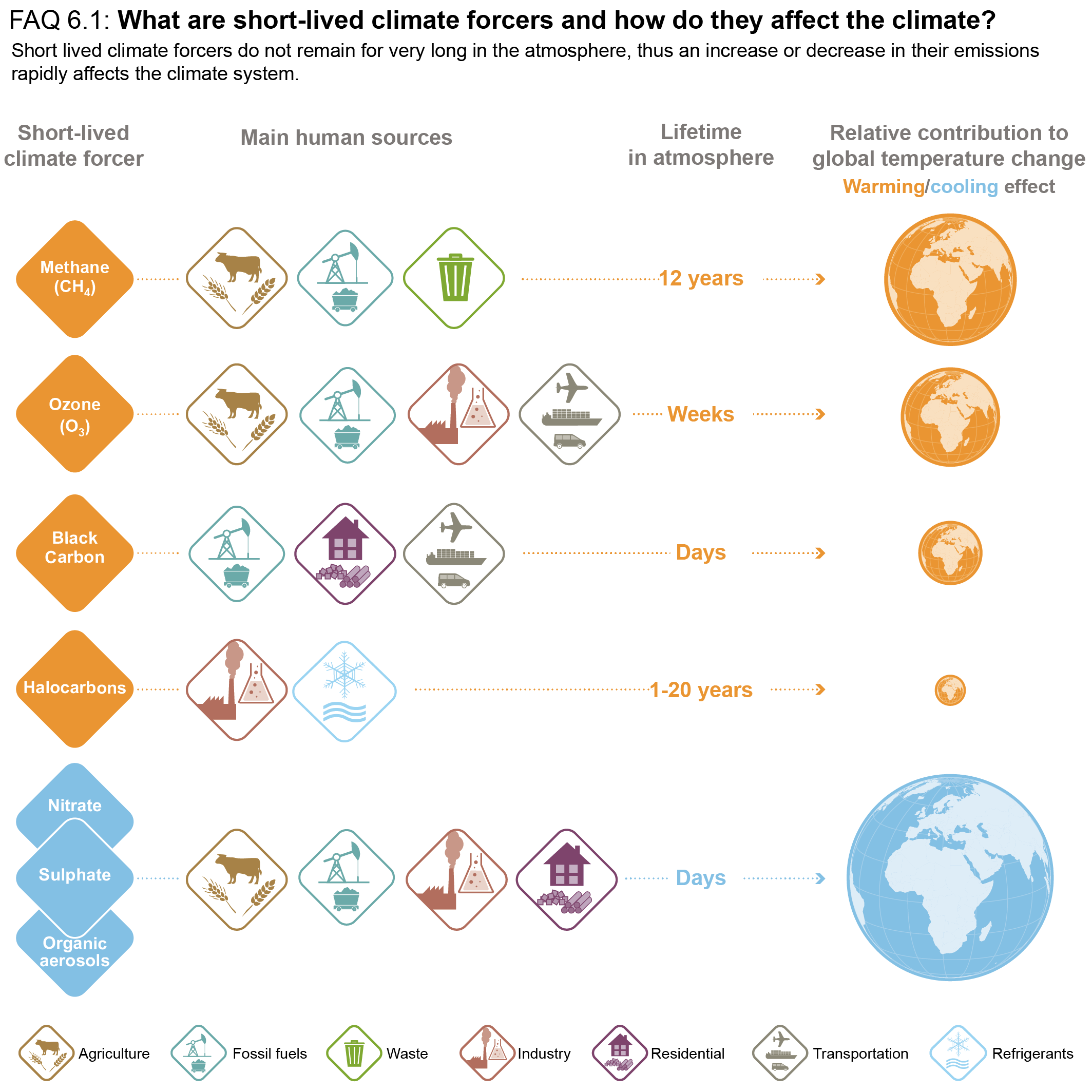
FAQ 6.1, Figure 1 | Main short-lived climate forcers, their sources, how long they exist in the atmosphere, and their relative contribution to global surface temperature changes between 1750 and 2019 (area of the globe). By definition this contribution depends on the lifetime, the warming/cooling potential (radiative efficiency), and the emissions of each compound in the atmosphere. Blue indicates cooling and orange indicates warming. Note that, between 1750 and 2019, the cooling contribution from aerosols (blue diamonds and globe) was approximately half the warming contribution from carbon dioxide.
FAQ 6.2 | What Are the Links Between Limiting Climate Change and Improving Air Quality?
Climate change and air quality are intimately linked. Many of the human activities that produce long-lived greenhouse gases also emit air pollutants, and many of these air pollutants are also ‘short-lived climate forcers’ that affect the climate. Therefore, many options for improving air quality may also serve to limit climate change and vice versa. However, some options for improving air quality cause additional climate warming, and some actions that address climate change can worsen air quality.
Climate change and air pollution are both critical environmental issues that are already affecting humanity. In 2016, the World Health Organization attributed 4.2 million deaths worldwide every year to ambient (outdoor) air pollution. Meanwhile, climate change impacts water resources, food production, human health, extreme events, coastal erosion, wildfires, and many other phenomena.
Most human activities, including energy production, agriculture, transportation, industrial processes, waste management and residential heating and cooling, result in emissions of gaseous and particulate pollutants that modify the composition of the atmosphere, leading to degradation of air quality as well as to climate change. These air pollutants are alsoshort-lived climate forcers– substances that affect the climate but remain in the atmosphere for shorter periods (days to decades) than long-lived greenhouse gases like carbon dioxide (see FAQ 6.1). While this means that the issues of air pollution and climate change are intimately connected, air pollutants and greenhouse gases are often defined, investigated and regulated independently of one another in both the scientific and policy arenas.
Many sources simultaneously emit carbon dioxide and air pollutants. When we drive our fossil fuel vehicles or light a fire in the fireplace, it is not just carbon dioxide or air pollutants that are emitted, but always both. It is therefore not possible to separate emissions into two clearly distinct groups. As a result, policies aiming at addressing climate change may have benefits or side effects for air quality, and vice versa.
For example, some short-term ‘win-win’ policies that simultaneously improve air quality and limit climate change include the implementation of energy efficiency measures, methane capture and recovery from solid-waste management and the oil and gas industry, zero-emissions vehicles, efficient and clean stoves for heating and cooking, filtering of soot (particulate matter) for diesel vehicles, cleaner brick-kiln technology, practices that reduce burning of agricultural waste, and the eradication of burning of kerosene for lighting.
There are, however, also ‘win-lose’ actions. For example, wood burning is defined as carbon neutral because a tree accumulates the same amount of carbon dioxide throughout its lifetime as is released when wood from that tree is burned. However, burning wood can also result in significant emissions of air pollutants, including carbon monoxide, nitrogen oxides, volatile organic compounds, and particulate matter, that locally or regionally affect the climate, human health and ecosystems (FAQ 6.2, Figure 1). Alternatively, decreasing the amount of sulphate aerosols produced by power and industrial plants, and from maritime transport, improves air quality but results in a warming influence on the climate, because those sulphate aerosols contribute to cooling the atmosphere by blocking incoming sunlight.
Air quality and climate change represent two sides of the same coin, and addressing both issues together could lead to significant synergies and economic benefits while avoiding policy actions that mitigate one of the two issues but worsen the other.
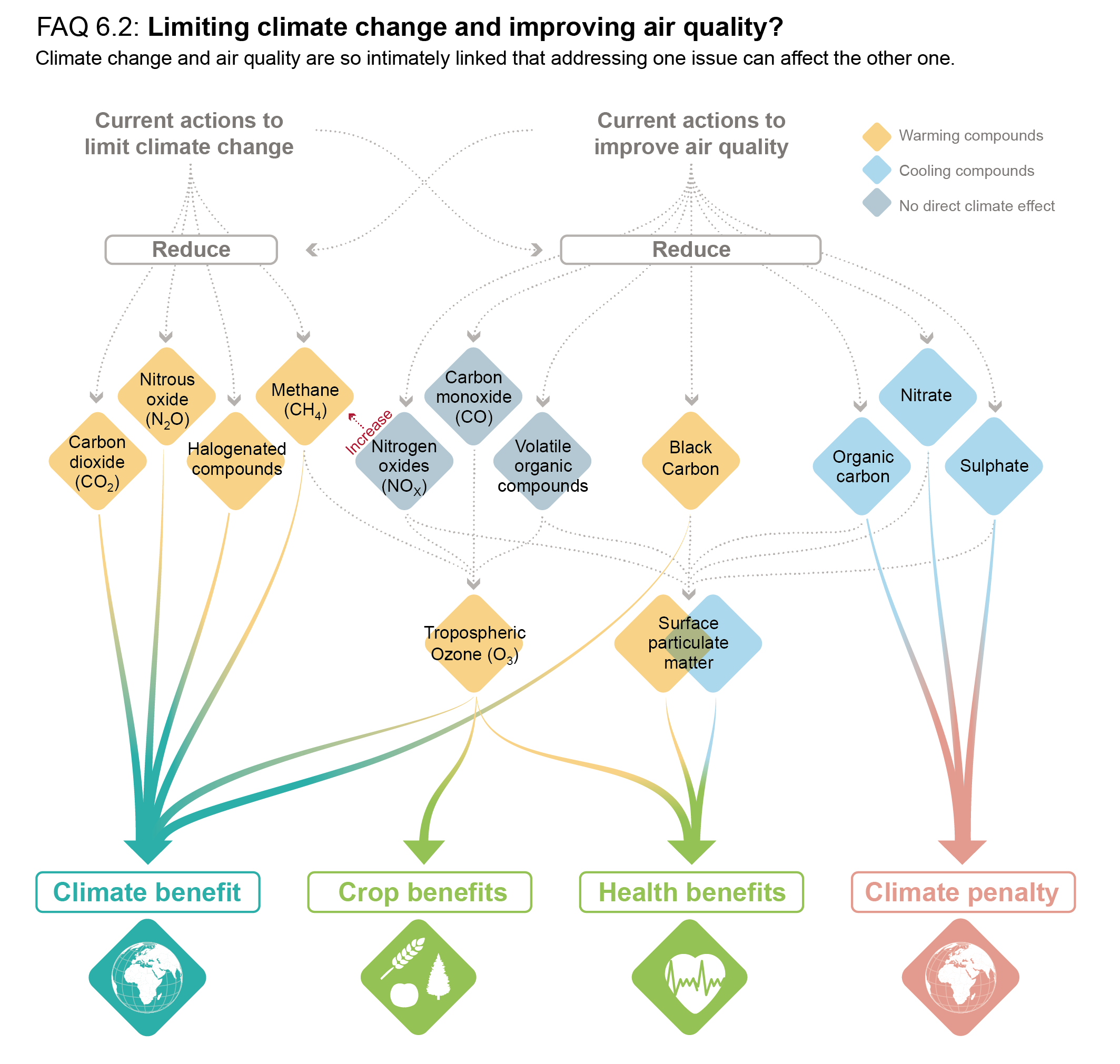
FAQ 6.2, Figure 1 | Links between actions aiming to limit climate change and actions to improve air quality. Greenhouse gases (GHGs) and aerosols (orange and blue) can affect climate directly. Air pollutants (bottom) can affect human health, ecosystems and climate. All these compounds have common sources and sometimes interact with each other in the atmosphere which makes it impossible to consider them separately (dotted grey arrows).
References
Aakre, S., S. Kallbekken, R. Van Dingenen, and D.G. Victor, 2018: Incentives for small clubs of Arctic countries to limit black carbon and methane emissions. Nature Climate Change, 8(1), 85–90, doi: 10.1038/s41558-017-0030-8.
Aamaas, B., T.K. Berntsen, J.S. Fuglestvedt, K.P. Shine, and N. Bellouin, 2016: Regional emission metrics for short-lived climate forcers from multiple models. Atmospheric Chemistry and Physics, 16(11), 7451–7468, doi: 10.5194/acp-16-7451-2016.
Aamaas, B., T.K. Berntsen, J.S. Fuglestvedt, K.P. Shine, and W.J. Collins, 2017: Regional temperature change potentials for short-lived climate forcers based on radiative forcing from multiple models. Atmospheric Chemistry and Physics, 17(17), 10795–10809, doi: 10.5194/acp-17-10795-2017.
Aas, W. et al., 2019: Global and regional trends of atmospheric sulfur. Scientific Reports, 9(1), 953, doi: 10.1038/s41598-018-37304-0.
Achakulwisut, P. et al., 2015: Uncertainties in isoprene photochemistry and emissions: Implications for the oxidative capacity of past and present atmospheres and for climate forcing agents. Atmospheric Chemistry and Physics, 15(14), 7977–7998, doi: 10.5194/acp-15-7977-2015.
Adebiyi, A.A. and J.F. Kok, 2020: Climate models miss most of the coarse dust in the atmosphere. Science Advances, 6(15), eaaz9507, doi: 10.1126/sciadv.aaz9507.
Aggarwal, S.G. and K. Kawamura, 2009: Carbonaceous and inorganic composition in long-range transported aerosols over northern Japan: Implication for aging of water-soluble organic fraction. Atmospheric Environment, 43(16), 2532–2540, doi: 10.1016/j.atmosenv.2009.02.032.
Ahlm, L. et al., 2017: Marine cloud brightening – as effective without clouds. Atmospheric Chemistry and Physics, 17(21), 13071–13087, doi: 10.5194/acp-17-13071-2017.
Ainsworth, E.A., C.R. Yendrek, S. Sitch, W.J. Collins, and L.D. Emberson, 2012: The Effects of Tropospheric Ozone on Net Primary Productivity and Implications for Climate Change. Annual Review of Plant Biology, 63(1), 637–661, doi: 10.1146/annurev-arplant-042110-103829.
Akagi, S.K. et al., 2011: Emission factors for open and domestic biomass burning for use in atmospheric models. Atmospheric Chemistry and Physics, 11(9), 4039–4072, doi: 10.5194/acp-11-4039-2011.
Akimoto, H. et al., 2015: SLCP co-control approach in East Asia: Tropospheric ozone reduction strategy by simultaneous reduction of NOx/NMVOC and methane. Atmospheric Environment, 122, 588–595, doi: 10.1016/j.atmosenv.2015.10.003.
Akritidis, D., A. Pozzer, and P. Zanis, 2019: On the impact of future climate change on tropopause folds and tropospheric ozone. Atmospheric Chemistry and Physics, 19(22), 14387–14401, doi: 10.5194/acp-19-14387-2019.
Alexander, B. and L.J. Mickley, 2015: Paleo-Perspectives on Potential Future Changes in the Oxidative Capacity of the Atmosphere Due to Climate Change and Anthropogenic Emissions. Current Pollution Reports, 1(2), 57–69, doi: 10.1007/s40726-015-0006-0.
Allen, D.J. and K.E. Pickering, 2002: Evaluation of lightning flash rate parameterizations for use in a global chemical transport model. Journal of Geophysical Research: Atmospheres, 107(D23), ACH 15-1-ACH 15-21, doi: 10.1029/2002jd002066.
Allen, D.J., K.E. Pickering, E. Bucsela, N. Krotkov, and R. Holzworth, 2019: Lightning NOx production in the Tropics as Determined Using OMI NO2 Retrievals and WWLLN Stroke Data. Journal of Geophysical Research: Atmospheres, 124(23), 13498–13518, doi: 10.1029/2018jd029824.
Allen, M.R. et al., 2016: New use of global warming potentials to compare cumulative and short-lived climate pollutants. Nature Climate Change, 6(8), 773–776, doi: 10.1038/nclimate2998.
Allen, M.R. et al., 2018a: Framing and Context. In: Global Warming of 1.5°C. An IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty[Masson-Delmotte, V., P. Zhai, H.-O. Pörtner, D. Roberts, J. Skea, P.R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J.B.R. Matthews, Y. Chen, X. Zhou, M.I. Gomis, E. Lonnoy, T. Maycock, M. Tignor, and T. Waterfield (eds.)]. In Press, pp. 49–92, www.ipcc.ch/sr15/chapter/chapter-1.
Allen, M.R. et al., 2018b: A solution to the misrepresentations of CO2 -equivalent emissions of short-lived climate pollutants under ambitious mitigation. npj Climate and Atmospheric Science, 1(1), 16, doi: 10.1038/s41612-018-0026-8.
Allen, R.J., W. Landuyt, and S.T. Rumbold, 2016: An increase in aerosol burden and radiative effects in a warmer world. Nature Climate Change, 6(3), 269–274, doi: 10.1038/nclimate2827.
Allen, R.J., T. Hassan, C.A. Randles, and H. Su, 2019: Enhanced land–sea warming contrast elevates aerosol pollution in a warmer world. Nature Climate Change, 9(4), 300–305, doi: 10.1038/s41558-019-0401-4.
Allen, R.J. et al., 2020: Climate and air quality impacts due to mitigation of non-methane near-term climate forcers. Atmospheric Chemistry and Physics, 20(16), 9641–9663, doi: 10.5194/acp-20-9641-2020.
Allen, R.J. et al., 2021: Significant climate benefits from near-term climate forcer mitigation in spite of aerosol reductions. Environmental Research Letters, 16(3), 034010, doi: 10.1088/1748-9326/abe06b.
Alterskjaer, K. and J.E. Kristjánsson, 2013: The sign of the radiative forcing from marine cloud brightening depends on both particle size and injection amount. Geophysical Research Letters, 40(1), 210–215, doi: 10.1029/2012gl054286.
Alvarez, R.A. et al., 2018: Assessment of methane emissions from the U.S. oil and gas supply chain. Science, 361(6398), 186–188, doi: 10.1126/science.aar7204.
Amador-Jiménez, M., N. Millner, C. Palmer, R.T. Pennington, and L. Sileci, 2020: The Unintended Impact of Colombia’s Covid-19 Lockdown on Forest Fires. Environmental and Resource Economics, 76(4), 1081–1105, doi: 10.1007/s10640-020-00501-5.
Amann, M., Z. Klimont, and F. Wagner, 2013: Regional and Global Emissions of Air Pollutants: Recent Trends and Future Scenarios. Annual Review of Environment and Resources, 38(1), 31-55, doi: 10.1146/annurev-environ-052912-173303.
Amann, M. et al., 2011: Cost-effective control of air quality and greenhouse gases in Europe: Modeling and policy applications. Environmental Modelling and Software, 26(12), 1489–1501, doi: 10.1016/j.envsoft.2011.07.012.
Amann, M. et al., 2017: Managing future air quality in megacities: A case study for Delhi. Atmospheric Environment, 161, 99–111, doi: 10.1016/j.atmosenv.2017.04.041.
Amann, M. et al., 2020: Reducing global air pollution: the scope for further policy interventions. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 378(2183), 20190331, doi: 10.1098/rsta.2019.0331.
AMAP, 2015a: AMAP Assessment 2015: Black carbon and ozone as Arctic climate forcers. Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, 116 pp., www.amap.no/documents/doc/amap-assessment-2015-black-carbon-and-ozone-as-arctic-climate-forcers/1299.
AMAP, 2015b: AMAP Assessment 2015: Methane as an Arctic climate forcer. Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, 139 pp., www.amap.no/documents/doc/amap-assessment-2015-methane-as-an-arctic-climate-forcer/1285.
Andrade, M.F. et al., 2017: Air quality in the megacity of São Paulo: Evolution over the last 30 years and future perspectives. Atmospheric Environment, 159, 66–82, doi: 10.1016/j.atmosenv.2017.03.051.
Andreae, M.O., 2019: Emission of trace gases and aerosols from biomass burning – An updated assessment. Atmospheric Chemistry and Physics, 19(13), 8523–8546, doi: 10.5194/acp-19-8523-2019.
Andreae, M.O. et al., 2015: The Amazon Tall Tower Observatory (ATTO): overview of pilot measurements on ecosystem ecology, meteorology, trace gases, and aerosols. Atmospheric Chemistry and Physics, 15(18), 10723–10776, doi: 10.5194/acp-15-10723-2015.
Andrews, A.E. et al., 2014: CO2, CO, and CH4 measurements from tall towers in the NOAA Earth System Research Laboratory’s Global Greenhouse Gas Reference Network: instrumentation, uncertainty analysis, and recommendations for future high-accuracy greenhouse gas monitoring efforts. Atmospheric Measurement Techniques, 7(2), 647–687, doi: 10.5194/amt-7-647-2014.
Anenberg, S.C., J. Miller, D. Henze, and R. Minjares, 2019: A global snapshot of the air pollution-related health impacts of transportation sector emissions in 2010 and 2015. International Council on Clean Transportation (ICCT), Washington, DC, USA, 55 pp., www.theicct.org/sites/default/files/publications/Global_health_impacts_transport_emissions_2010-2015_20190226.pdf .
Anenberg, S.C. et al., 2012: Global air quality and health co-benefits of mitigating near-term climate change through methane and black carbon emission controls. Environmental Health Perspectives, 120(6), 831–839, doi: 10.1289/ehp.1104301.
Anenberg, S.C. et al., 2017: Impacts and mitigation of excess diesel-related NOx emissions in 11 major vehicle markets. Nature, 545(7655), 467–471, doi: 10.1038/nature22086.
Angelbratt, J. et al., 2011: Carbon monoxide (CO) and ethane (C2 h6) trends from ground-based solar FTIR measurements at six European stations, comparison and sensitivity analysis with the EMEP model. Atmospheric Chemistry and Physics, 11(17), 9253–9269, doi: 10.5194/acp-11-9253-2011.
Archibald, A.T. et al., 2020: Tropospheric Ozone Assessment Report: A critical review of changes in the tropospheric ozone burden and budget from 1850 to 2100. Elementa: Science of the Anthropocene, 8(1), doi: 10.1525/elementa.2020.034.
Arneth, A., R.K. Monson, G. Schurgers, Niinemets, and P.I. Palmer, 2008: Why are estimates of global terrestrial isoprene emissions so similar (and why is this not so for monoterpenes)?Atmospheric Chemistry and Physics, 8(16), 4605–4620, doi: 10.5194/acp-8-4605-2008.
Arneth, A. et al., 2010a: Terrestrial biogeochemical feedbacks in the climate system. Nature Geoscience, 3(8), 525–532, doi: 10.1038/ngeo905.
Arneth, A. et al., 2010b: From biota to chemistry and climate: towards a comprehensive description of trace gas exchange between the biosphere and atmosphere. Biogeosciences, 7(1), 121–149, doi: 10.5194/bg-7- 121-2010.
Arnold, S.R. et al., 2018: Simulated Global Climate Response to Tropospheric Ozone-Induced Changes in Plant Transpiration. Geophysical Research Letters, 45(23), 13,13–70,79, doi: 10.1029/2018gl079938.
Aschmann, J. et al., 2014: On the hiatus in the acceleration of tropical upwelling since the beginning of the 21st century. Atmospheric Chemistry and Physics, 14(23), 12803–12814, doi: 10.5194/acp-14-12803-2014.
Athanasopoulou, E. et al., 2017: Changes in domestic heating fuel use in Greece: effects on atmospheric chemistry and radiation. Atmospheric Chemistry and Physics, 17(17), 10597–10618, doi: 10.5194/acp-17-10597-2017.
Baker, L.H. et al., 2015: Climate responses to anthropogenic emissions of short-lived climate pollutants. Atmospheric Chemistry and Physics, 15(14), 8201–8216, doi: 10.5194/acp-15-8201-2015.
Balkanski, Y. et al., 2010: Direct radiative effect of aerosols emitted by transport: from road, shipping and aviation. Atmospheric Chemistry and Physics, 10(10), 4477–4489, doi: 10.5194/acp-10-4477-2010.
Banerjee, A. et al., 2016: Drivers of changes in stratospheric and tropospheric ozone between year 2000 and 2100. Atmospheric Chemistry and Physics, 16(5), 2727–2746, doi: 10.5194/acp-16-2727-2016.
Banzhaf, S. et al., 2015: Dynamic model evaluation for secondary inorganic aerosol and its precursors over Europe between 1990 and 2009. Geoscientific Model Development, 8(4), 1047–1070, doi: 10.5194/gmd-8-1047-2015.
Barkley, M.P. et al., 2013: Top-down isoprene emissions over tropical South America inferred from SCIAMACHY and OMI formaldehyde columns. Journal of Geophysical Research: Atmospheres, 118(12), 6849–6868, doi: 10.1002/jgrd.50552.
Barnes, E.A., A.M. Fiore, and L.W. Horowitz, 2016: Detection of trends in surface ozone in the presence of climate variability. Journal of Geophysical Research: Atmospheres, 121(10), 6112–6129, doi: 10.1002/2015jd024397.
Barthel, S., I. Tegen, and R. Wolke, 2019: Do new sea spray aerosol source functions improve the results of a regional aerosol model?Atmospheric Environment, 198, 265–278, doi: 10.1016/j.atmosenv.2018.10.016.
Batmunkh, T. et al., 2011: Time-Resolved Measurements of PM2.5 Carbonaceous Aerosols at Gosan, Korea. Journal of the Air & Waste Management Association, 61(11), 1174–1182, doi: 10.1080/10473289.2011.609761.
Bauer, S.E., K. Tsigaridis, and R. Miller, 2016: Significant atmospheric aerosol pollution caused by world food cultivation. Geophysical Research Letters, 43(10), 5394–5400, doi: 10.1002/2016gl068354.
Bauer, S.E. et al., 2020: Historical (1850–2014) Aerosol Evolution and Role on Climate Forcing Using the GISS ModelE2.1 Contribution to CMIP6. Journal of Advances in Modeling Earth Systems, 12(8), e2019MS001978, doi: 10.1029/2019ms001978.
Bauwens, M. et al., 2020: Impact of Coronavirus Outbreak on NO2 pollution Assessed Using TROPOMI and OMI Observations. Geophysical Research Letters, 47(11), doi: 10.1029/2020gl087978.
Beekmann, M. et al., 2015: In situ, satellite measurement and model evidence on the dominant regional contribution to fine particulate matter levels in the Paris megacity. Atmospheric Chemistry and Physics, 15, 9577–9591, doi: 10.5194/acp-15-9577-2015.
Bekki, S. and F. Lefevre, 2009: Stratospheric ozone: History and concepts and interactions with climate. EPJ Web of Conferences, 1, 113–136, doi: 10.1140/epjconf/e2009-00914-y.
Bekki, S. et al., 2013: Climate impact of stratospheric ozone recovery. Geophysical Research Letters, 40(11), 2796–2800, doi: 10.1002/grl.50358.
Bellouin, N. et al., 2013: Impact of the modal aerosol scheme GLOMAP-mode on aerosol forcing in the Hadley Centre Global Environmental Model. Atmospheric Chemistry and Physics, 13(6), 3027–3044, doi: 10.5194/acp-13-3027-2013.
Bellouin, N. et al., 2020: Bounding Global Aerosol Radiative Forcing of Climate Change. Reviews of Geophysics, 58(1), e2019RG000660, doi: 10.1029/2019rg000660.
Berntsen, T. and J. Fuglestvedt, 2008: Global temperature responses to current emissions from the transport sectors. Proceedings of the National Academy of Sciences, 105(49), 19154–19159, doi: 10.1073/pnas.0804844105.
Bhaskar, V.V. and P.S.P. Rao, 2017: Annual and decadal variation in chemical composition of rain water at all the ten GAW stations in India. Journal of Atmospheric Chemistry, 74(1), 23–53, doi: 10.1007/s10874-016-9339-3.
Bian, H. et al., 2017: Investigation of global particulate nitrate from the AeroCom phase III experiment. Atmospheric Chemistry and Physics, 17(21), 12911–12940, doi: 10.5194/acp-17-12911-2017.
Bickel, M., M. Ponater, L. Bock, U. Burkhardt, and S. Reineke, 2020: Estimating the Effective Radiative Forcing of Contrail Cirrus. Journal of Climate, 33(5), 1991–2005, doi: 10.1175/jcli-d-19-0467.1.
Bishop, G.A. and M.J. Haugen, 2018: The Story of Ever Diminishing Vehicle Tailpipe Emissions as Observed in the Chicago, Illinois Area. Environmental Science & Technology, 52(13), 7587–7593, doi: 10.1021/acs.est.8b00926.
Bond, T.C. et al., 2013: Bounding the role of black carbon in the climate system: A scientific assessment. Journal of Geophysical Research: Atmospheres, 118(11), 5380–5552, doi: 10.1002/jgrd.50171.
Bondur, V.G., I.I. Mokhov, O.S. Voronova, and S.A. Sitnov, 2020: Satellite Monitoring of Siberian Wildfires and Their Effects: Features of 2019 Anomalies and Trends of 20-Year Changes. Doklady Earth Sciences, 492(1), 370–375, doi: 10.1134/s1028334x20050049.
Bonjour, S. et al., 2013: Solid fuel use for household cooking: Country and regional estimates for 1980–2010. Environmental Health Perspectives, 121(7), 784–790, doi: 10.1289/ehp.1205987.
Bopp, L. et al., 2004: Will marine dimethylsulfide emissions amplify or alleviate global warming? A model study. Canadian Journal of Fisheries and Aquatic Sciences, 61(5), 826–835, doi: 10.1139/f04-045.
Boucher, O. et al., 2013: Clouds and Aerosols. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change[Stocker, T.F., D. Qin, G.-K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P.M. Midgley (eds.)]. Cambridge University Press, Cambridge, pp. 571–658, doi: 10.1017/cbo9781107415324.016.
Bourotte, C. et al., 2007: Association between ionic composition of fine and coarse aerosol soluble fraction and peak expiratory flow of asthmatic patients in São Paulo city (Brazil). Atmospheric Environment, 41(10), 2036–2048, doi: 10.1016/j.atmosenv.2006.11.004.
Bowman, D.M.J.S. et al., 2009: Fire in the earth system. Science, 324(5926), 481–484, doi: 10.1126/science.1163886.
Braspenning Radu, O. et al., 2016: Exploring synergies between climate and air quality policies using long-term global and regional emission scenarios. Atmospheric Environment, 140, 577–591, doi: 10.1016/j.atmosenv.2016.05.021.
Brasseur, G.P. and D.J. Jacob, 2017: Modeling of Atmospheric Chemistry. Cambridge University Press, UK, 606 pp., doi: 10.1017/9781316544754.
Breider, T.J. et al., 2017: Multidecadal trends in aerosol radiative forcing over the Arctic: Contribution of changes in anthropogenic aerosol to Arctic warming since 1980. Journal of Geophysical Research: Atmospheres, 122(6), 3573–3594, doi: 10.1002/2016jd025321.
Brown-Steiner, B. et al., 2018: Evaluating simplified chemical mechanisms within present-day simulations of the Community Earth System Model version 1.2 with CAM4 (CESM1.2 CAM-chem): MOZART-4 vs. Reduced Hydrocarbon vs. Super-Fast chemistry. Geoscientific Model Development, 11(10), 4155–4174, doi: 10.5194/gmd-11-4155-2018.
Buchholz, R.R. et al., 2021: Air pollution trends measured from Terra: CO and AOD over industrial, fire-prone, and background regions. Remote Sensing of Environment, 256, 112275, doi: 10.1016/j.rse.2020.112275.
Bucsela, E.J., K.E. Pickering, D.J. Allen, R.H. Holzworth, and N.A. Krotkov, 2019: Midlatitude Lightning NOx production Efficiency Inferred From OMI and WWLLN Data. Journal of Geophysical Research: Atmospheres, 124(23), 13475–13497, doi: 10.1029/2019jd030561.
Bunzel, F. and H. Schmidt, 2013: The Brewer-Dobson Circulation in a Changing Climate: Impact of the Model Configuration. Journal of the Atmospheric Sciences, 70(5), 1437–1455, doi: 10.1175/jas-d-12-0215.1.
Butler, T., F. Vermeylen, C.M. Lehmann, G.E. Likens, and M. Puchalski, 2016: Increasing ammonia concentration trends in large regions of the USA derived from the NADP/AMoN network. Atmospheric Environment, 146, 132–140, doi: 10.1016/j.atmosenv.2016.06.033.
Butt, E.W. et al., 2016: The impact of residential combustion emissions on atmospheric aerosol, human health, and climate. Atmospheric Chemistry and Physics, 16(2), 873–905, doi: 10.5194/acp-16-873-2016.
Cai, W., K. Li, H. Liao, H. Wang, and L. Wu, 2017: Weather conditions conducive to Beijing severe haze more frequent under climate change. Nature Climate Change, 7(4), 257–262, doi: 10.1038/nclimate3249.
Caldeira, K., G. Bala, and L. Cao, 2013: The Science of Geoengineering. Annual Review of Earth and Planetary Sciences, 41(1), 231–256, doi: 10.1146/annurev-earth-042711-105548.
Callaghan, A.H., M.D. Stokes, and G.B. Deane, 2014: The effect of water temperature on air entrainment, bubble plumes, and surface foam in a laboratory breaking-wave analog. Journal of Geophysical Research: Oceans, 119(11), 7463–7482, doi: 10.1002/2014jc010351.
Carpenter, L.J. et al., 2014: Update on Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol, Chapter 1. In: Scientific Assessment of Ozone Depletion: 2014. Global Ozone Research and Monitoring Project – Report No. 55, World Meteorological Organization (WMO), Geneva, Switzerland, pp. 1.1–1.101, https://csl.noaa.gov/assessments/ozone/2014/report/.
Carslaw, K.S. et al., 2010: A review of natural aerosol interactions and feedbacks within the Earth system. Atmospheric Chemistry and Physics, 10(4), 1701–1737, doi: 10.5194/acp-10-1701-2010.
Cecil, D.J., D.E. Buechler, and R.J. Blakeslee, 2014: Gridded lightning climatology from TRMM-LIS and OTD: Dataset description. Atmospheric Research, 135–136, 404–414, doi: 10.1016/j.atmosres.2012.06.028.
Celis, J.E., J.R. Morales, C.A. Zaror, and J.C. Inzunza, 2004: A study of the particulate matter PM10 composition in the atmosphere of Chillán, Chile. Chemosphere, 54(4), 541–550, doi: 10.1016/s0045-6535(03)00711-2.
Chambliss, S.E., R. Silva, J.J. West, M. Zeinali, and R. Minjares, 2014: Estimating source-attributable health impacts of ambient fine particulate matter exposure: Global premature mortality from surface transportation emissions in 2005. Environmental Research Letters, 9(10), 104009, doi: 10.1088/1748-9326/9/10/104009.
Chang, K.M. et al., 2017: Ancillary health effects of climate mitigation scenarios as drivers of policy uptake: A review of air quality, transportation and diet co-benefits modeling studies. Environmental Research Letters, 12(11), 113001, doi: 10.1088/1748-9326/aa8f7b.
Chen, D., Z. Liu, J. Fast, and J. Ban, 2016: Simulations of sulfate-nitrate-ammonium (SNA) aerosols during the extreme haze events over northern China in October 2014. Atmospheric Chemistry and Physics, 16(16), 10707–10724, doi: 10.5194/acp-16-10707-2016.
Chen, K. et al., 2018: Future ozone-related acute excess mortality under climate and population change scenarios in China: A modeling study. PLOS Medicine, 15(7), e1002598, doi: 10.1371/journal.pmed.1002598.
Chen, S. et al., 2017: Direct observations of organic aerosols in common wintertime hazes in North China: Insights into direct emissions from Chinese residential stoves. Atmospheric Chemistry and Physics, 17(2), 1259–1270, doi: 10.5194/acp-17-1259-2017.
Cheng, Y. et al., 2016: Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China. Science Advances, 2(12), e1601530, doi: 10.1126/sciadv.1601530.
Cherian, R. and J. Quaas, 2020: Trends in AOD, Clouds, and Cloud Radiative Effects in Satellite Data and CMIP5 and CMIP6 Model Simulations Over Aerosol Source Regions. Geophysical Research Letters, 47(9), e2020GL087132, doi: 10.1029/2020gl087132.
Chevallier, F. et al., 2020: Local Anomalies in the Column-Averaged Dry Air Mole Fractions of Carbon Dioxide Across the Globe During the First Months of the Coronavirus Recession. Geophysical Research Letters, 47(22), e2020GL090244, doi: 10.1029/2020gl090244.
Cho, S.Y. and S.S. Park, 2013: Resolving sources of water-soluble organic carbon in fine particulate matter measured at an urban site during winter. Environmental Science: Processes & Impacts, 15, 524–534, doi: 10.1039/c2em30730h.
Cholakian, A., A. Colette, I. Coll, G. Ciarelli, and M. Beekmann, 2019: Future climatic drivers and their effect on PM10 components in Europe and the Mediterranean Sea. Atmospheric Chemistry and Physics, 19(7), 4459–4484, doi: 10.5194/acp-19-4459-2019.
Chossière, G.P. et al., 2018: Country- and manufacturer-level attribution of air quality impacts due to excess NOx emissions from diesel passenger vehicles in Europe. Atmospheric Environment, 189, 89–97, doi: 10.1016/j.atmosenv.2018.06.047.
Christensen, T.R., V.K. Arora, M. Gauss, L. Höglund-Isaksson, and F.J.W. Parmentier, 2019: Tracing the climate signal: mitigation of anthropogenic methane emissions can outweigh a large Arctic natural emission increase. Scientific Reports, 9(1), 1146, doi: 10.1038/s41598-018-37719-9.
Chrysanthou, A. et al., 2019: The effect of atmospheric nudging on the stratospheric residual circulation in chemistry–climate models. Atmospheric Chemistry and Physics, 19(17), 11559–11586, doi: 10.5194/acp-19-11559-2019.
Chuvieco, E. et al., 2016: A new global burned area product for climate assessment of fire impacts. Global Ecology and Biogeography, 25(5), 619–629, doi: 10.1111/geb.12440.
Chuvieco, E. et al., 2019: Historical background and current developments for mapping burned area from satellite Earth observation. Remote Sensing of Environment, 225, 45–64, doi: 10.1016/j.rse.2019.02.013.
Chuwah, C. et al., 2013: Implications of alternative assumptions regarding future air pollution control in scenarios similar to the Representative Concentration Pathways. Atmospheric Environment, 79, 787–801, doi: 10.1016/j.atmosenv.2013.07.008.
Ciais, P. et al., 2013: Carbon and Other Biogeochemical Cycles. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change[Stocker, T.F., D. Qin, G.-K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P.M. Midgley (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 465–570, doi: 10.1017/cbo9781107415324.015.
Cirino, G.G., R.A.F. Souza, D.K. Adams, and P. Artaxo, 2014: The effect of atmospheric aerosol particles and clouds on net ecosystem exchange in the Amazon. Atmospheric Chemistry and Physics, 14(13), 6523–6543, doi: 10.5194/acp-14-6523-2014.
Clarisse, L., C. Clerbaux, F. Dentener, D. Hurtmans, and P.F. Coheur, 2009: Global ammonia distribution derived from infrared satellite observations. Nature Geoscience, 2(7), 479–483, doi: 10.1038/ngeo551.
Clark, S.K., D.S. Ward, and N.M. Mahowald, 2017: Parameterization-based uncertainty in future lightning flash density. Geophysical Research Letters, 44(6), 2893–2901, doi: 10.1002/2017gl073017.
Cochran, R.E., O.S. Ryder, V.H. Grassian, and K.A. Prather, 2017: Sea Spray Aerosol: The Chemical Link between the Oceans, Atmosphere, and Climate. Accounts of Chemical Research, 50(3), 599–604, doi: 10.1021/acs.accounts.6b00603.
Cohen, Y. et al., 2018: Climatology and long-term evolution of ozone and carbon monoxide in the upper troposphere-lower stratosphere (UTLS) at northern midlatitudes, as seen by IAGOS from 1995 to 2013. Atmospheric Chemistry and Physics, 18(8), 5415–5453, doi: 10.5194/acp-18-5415-2018.
Colette, A. et al., 2015: Is the ozone climate penalty robust in Europe?Environmental Research Letters, 10(8), 084015, doi: 10.1088/1748-9326/10/8/084015.
Collins, W.J., S. Sitch, and O. Boucher, 2010: How vegetation impacts affect climate metrics for ozone precursors. Journal of Geophysical Research: Atmospheres, 115(D23), D23308, doi: 10.1029/2010jd014187.
Collins, W.J. et al., 2013: Global and regional temperature-change potentials for near-term climate forcers. Atmospheric Chemistry and Physics, 13(5), 2471–2485, doi: 10.5194/acp-13-2471-2013.
Collins, W.J. et al., 2017: AerChemMIP: Quantifying the effects of chemistry and aerosols in CMIP6. Geoscientific Model Development, 10(2), 585–607, doi: 10.5194/gmd-10-585-2017.
Collins, W.J. et al., 2018: Increased importance of methane reduction for a 1.5 degree target. Environmental Research Letters, 13(5), 54003, doi: 10.1088/1748-9326/aab89c.
Coninck, H. et al., 2018: Strengthening and Implementing the Global Response. In: Global warming of 1.5°C. An IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty[Masson-Delmotte, V., P. Zhai, H.O. Pörtner, D. Roberts, J. Skea, P.R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J.B.R. Matthews, Y. Chen, X. Zhou, M.I. Gomis, E. Lonnoy, T. Maycock, M. Tignor, and T. Waterfield (eds.)]. In Press, pp. 313–443, www.ipcc.ch/sr15/chapter/chapter-4.
Conrad, B.M. and M.R. Johnson, 2017: Field Measurements of Black Carbon Yields from Gas Flaring. Environmental Science & Technology, 51(3), 1893–1900, doi: 10.1021/acs.est.6b03690.
Cooper, O.R. et al., 2020: Multi-decadal surface ozone trends at globally distributed remote locations. Elementa: Science of the Anthropocene, 8(1), 23, doi: 10.1525/elementa.420.
Corbett, J.J. et al., 2007: Mortality from Ship Emissions: A Global Assessment. Environmental Science & Technology, 41(24), 8512–8518, doi: 10.1021/es071686z.
Cox, P.M. et al., 2008: Increasing risk of Amazonian drought due to decreasing aerosol pollution. Nature, 453(7192), 212–215, doi: 10.1038/nature06960.
Cravigan, L.T. et al., 2020: Sea spray aerosol organic enrichment, water uptake and surface tension effects. Atmospheric Chemistry and Physics, 20(13), 7955–7977, doi: 10.5194/acp-20-7955-2020.
Crippa, M. et al., 2016: Forty years of improvements in European air quality: regional policy-industry interactions with global impacts. Atmospheric Chemistry and Physics, 16(6), 3825–3841, doi: 10.5194/acp-16-3825-2016.
Crippa, M. et al., 2018: Gridded emissions of air pollutants for the period 1970–2012 within EDGAR v4.3.2. Earth System Science Data, 10(4), 1987–2013, doi: 10.5194/essd-10-1987-2018.
Crippa, M. et al., 2019: Fossil CO2and GHG emissions of all world countries – 2019 Report. JRC117610, Publications Office of the European Union, Luxembourg, 251 pp., doi: 10.2760/687800.
Crippa, M. et al., 2020: Fossil CO2 emissions of all world countries – 2020 Report. JRC121460, Publications Office of the European Union, Luxembourg, 244 pp., doi: 10.2760/143674.
Dai, Z., D.K. Weisenstein, and D.W. Keith, 2018: Tailoring Meridional and Seasonal Radiative Forcing by Sulfate Aerosol Solar Geoengineering. Geophysical Research Letters, 45(2), 1030–1039, doi: 10.1002/2017gl076472.
Dalsøren, S.B. et al., 2016: Atmospheric methane evolution the last 40 years. Atmospheric Chemistry and Physics, 16(5), 3099–3126, doi: 10.5194/acp-16-3099-2016.
Dalsøren, S.B. et al., 2018: Discrepancy between simulated and observed ethane and propane levels explained by underestimated fossil emissions. Nature Geoscience, 11(3), 178–184, doi: 10.1038/s41561-018-0073-0.
Dameris, M. and P. Jöckel, 2013: Numerical modeling of climate–chemistry connections: Recent developments and future challenges. Atmosphere, 4(2), 132–156, doi: 10.3390/atmos4020132.
Dang, R. and H. Liao, 2019: Radiative Forcing and Health Impact of Aerosols and Ozone in China as the Consequence of Clean Air Actions over 2012–2017. Geophysical Research Letters, 46(21), 12511–12519, doi: 10.1029/2019gl084605.
de Foy, B., Z. Lu, and D.G. Streets, 2016: Satellite NO2 retrievals suggest China has exceeded its NOx reduction goals from the twelfth Five-Year Plan. Scientific Reports, 6(1), 35912, doi: 10.1038/srep35912.
De Smedt, I. et al., 2015: Diurnal, seasonal and long-term variations of global formaldehyde columns inferred from combined OMI and GOME-2 observations. Atmospheric Chemistry and Physics, 15(21), 12519–12545, doi: 10.5194/acp-15-12519-2015.
de Souza, P.A., W.Z. Mello, R.L. Mariani, and S.M. Sella, 2010: Caracterização do material particulado fino e grosso e composição da fração inorgânica solúvel em água em São José dos Campos (SP). Quimica Nova, 33(6), 1247–1253, doi: 10.1590/s0100-40422010000600005.
de Vries, W., M. Posch, D. Simpson, and G.J. Reinds, 2017: Modelling long-term impacts of changes in climate, nitrogen deposition and ozone exposure on carbon sequestration of European forest ecosystems. Science of The Total Environment, 605–606, 1097–1116, doi: 10.1016/j.scitotenv.2017.06.132.
Deeter, M.N. et al., 2017: A climate-scale satellite record for carbon monoxide: The MOPITT Version 7 product. Atmospheric Measurement Techniques, 10(7), 2533–2555, doi: 10.5194/amt-10-2533-2017.
DeMott, P.J. et al., 2016: Sea spray aerosol as a unique source of ice nucleating particles. Proceedings of the National Academy of Sciences, 113(21), 5797–5803, doi: 10.1073/pnas.1514034112.
Dhaka, S.K. et al., 2020: PM2.5 diminution and haze events over Delhi during the COVID-19 lockdown period: an interplay between the baseline pollution and meteorology. Scientific Reports, 10(1), 13442, doi: 10.1038/s41598-020-70179-8.
Dietmüller, S., M. Ponater, and R. Sausen, 2014: Interactive ozone induces a negative feedback in CO2 -driven climate change simulations. Journal of Geophysical Research: Atmospheres, 119(4), 1796–1805, doi: 10.1002/2013jd020575.
Ding, S. et al., 2019: Observed Interactions Between Black Carbon and Hydrometeor During Wet Scavenging in Mixed-Phase Clouds. Geophysical Research Letters, 46(14), 8453–8463, doi: 10.1029/2019gl083171.
Dlugokencky, E. and P. Tans, 2021: Trends in Atmospheric Carbon Dioxide, Methane and Nitrous Oxide. Global Monitoring Laboratory, National Oceanic & Atmospheric Administration Earth System Research Laboratories (NOAA/ESRL). Retrieved from: www.esrl.noaa.gov/gmd/ccgg/trends.
Duncan, B.N. et al., 2013: The observed response of Ozone Monitoring Instrument (OMI) NO2 columns to NOx emission controls on power plants in the United States: 2005–2011. Atmospheric Environment, 81, 102–111, doi: 10.1016/j.atmosenv.2013.08.068.
Duncan, B.N. et al., 2014: Satellite data of atmospheric pollution for U.S. air quality applications: Examples of applications, summary of data end-user resources, answers to FAQs, and common mistakes to avoid. Atmospheric Environment, 94, 647–662, doi: 10.1016/j.atmosenv.2014.05.061.
Duncan, B.N. et al., 2016: A space-based, high-resolution view of notable changes in urban NOx pollution around the world (2005–2014). Journal of Geophysical Research: Atmospheres, 121(2), 976–996, doi: 10.1002/2015jd024121.
Dutkiewicz, V.A. et al., 2014: Forty-seven years of weekly atmospheric black carbon measurements in the Finnish Arctic: Decrease in black carbon with declining emissions. Journal of Geophysical Research: Atmospheres, 119(12), 7667–7683, doi: 10.1002/2014jd021790.
Dykema, J.A., D.W. Keith, and F.N. Keutsch, 2016: Improved aerosol radiative properties as a foundation for solar geoengineering risk assessment. Geophysical Research Letters, 43(14), 7758–7766, doi: 10.1002/2016gl069258.
EC-JRC/PBL, 2020: EDGAR v5.0 Global Air Pollutant Emissions. European Commission, Joint Research Centre (EC-JRC)/Netherlands Environmental Assessment Agency (PBL). Retrieved from: https://edgar.jrc.ec.europa.eu/overview.php?v=50_ap.
Ekman, A.M.L., 2014: Do sophisticated parameterizations of aerosol–cloud interactions in CMIP5 models improve the representation of recent observed temperature trends?Journal of Geophysical Research: Atmospheres, 119(2), 817–832, doi: 10.1002/2013jd020511.
Elguindi, N. et al., 2020: Intercomparison of Magnitudes and Trends in Anthropogenic Surface Emissions From Bottom-Up Inventories, Top-Down Estimates, and Emission Scenarios. Earth’s Future, 8(8), e2020EF001520, doi: 10.1029/2020ef001520.
Elvidge, E.C.L., S.-M. Phang, W.T. Sturges, and G. Malin, 2015a: The effect of desiccation on the emission of volatile bromocarbons from two common temperate macroalgae. Biogeosciences, 12(2), 387–398, doi: 10.5194/bg-12-387-2015.
Elvidge, E.C.L. et al., 2015b: Increasing concentrations of dichloromethane, CH2 cl2, inferred from CARIBIC air samples collected 1998–2012. Atmospheric Chemistry and Physics, 15(4), 1939–1958, doi: 10.5194/acp-15-1939-2015.
Emberson, L.D. et al., 2018: Ozone effects on crops and consideration in crop models. European Journal of Agronomy, 100, 19–34, doi: 10.1016/j.eja.2018.06.002.
Emmerson, K.M. and M.J. Evans, 2009: Comparison of tropospheric gas-phase chemistry schemes for use within global models. Atmospheric Chemistry and Physics, 9(5), 1831–1845, doi: 10.5194/acp-9-1831-2009.
Emmons, L.K. et al., 2020: The Chemistry Mechanism in the Community Earth System Model Version 2 (CESM2). Journal of Advances in Modeling Earth Systems, 12(4), e2019MS001882, doi: 10.1029/2019ms001882.
Engardt, M., D. Simpson, M. Schwikowski, and L. Granat, 2017: Deposition of sulphur and nitrogen in Europe 1900–2050. Model calculations and comparison to historical observations. Tellus B: Chemical and Physical Meteorology, 69(1), 1328945, doi: 10.1080/16000889.2017.1328945.
Engel, A. et al., 2018: Update on Ozone-Depleting Substances (ODSs) and Other Gases of Interest to the Montreal Protocol. In: Scientific Assessment of Ozone Depletion: 2018. Global Ozone Research and Monitoring Project – Report No. 58, World Meteorological Organization (WMO), Geneva, Switzerland, pp. 1.1–1.87, https://csl.noaa.gov/assessments/ozone/2018/downloads/.
EPA, 2019: Global Non-CO2Greenhouse Gas Emission Projections & Mitigation: 2015–2020. EPA-430-R-19-010, United States Environmental Protection Agency (EPA), Office of Atmospheric Programs (6207A), Washington, DC, USA, 84 pp., www.epa.gov/sites/production/files/2019-09/documents/epa_non-co2_greenhouse_gases_rpt-epa430r19010.pdf .
Erisman, J.W., M.A. Sutton, J. Galloway, Z. Klimont, and W. Winiwarter, 2008: How a century of ammonia synthesis changed the world. Nature Geoscience, 1(10), 636–639, doi: 10.1038/ngeo325.
Etiope, G. and P. Ciccioli, 2009: Earth’s degassing: A missing ethane and propane source. Science, 323(5913), 478, doi: 10.1126/science.1165904.
Evan, A.T., C. Flamant, S. Fiedler, and O. Doherty, 2014: An analysis of aeolian dust in climate models. Geophysical Research Letters, 41(16), 5996–6001, doi: 10.1002/2014gl060545.
Evans, M. et al., 2017: Black carbon emissions in Russia: A critical review. Atmospheric Environment, 163, 9–21, doi: 10.1016/j.atmosenv.2017.05.026.
Evans, S., P. Ginoux, S. Malyshev, and E. Shevliakova, 2016: Climate-vegetation interaction and amplification of Australian dust variability. Geophysical Research Letters, 43(22), 11823–11830, doi: 10.1002/2016gl071016.
Eyring, V. et al., 2010: Transport impacts on atmosphere and climate: Shipping. Atmospheric Environment, 44(37), 4735–4771, doi: 10.1016/j.atmosenv.2009.04.059.
Fanourgakis, G.S. et al., 2019: Evaluation of global simulations of aerosol particle and cloud condensation nuclei number, with implications for cloud droplet formation. Atmospheric Chemistry and Physics, 19(13), 8591–8617, doi: 10.5194/acp-19-8591-2019.
Favez, O. et al., 2008: Seasonality of major aerosol species and their transformations in Cairo megacity. Atmospheric Environment, 42(7), 1503–1516, doi: 10.1016/j.atmosenv.2007.10.081.
Fay, M. et al., 2018: Energy investment needs for fulfilling the Paris Agreement and achieving the Sustainable Development Goals. Nature Energy, 3(7), 589–599, doi: 10.1038/s41560-018-0179-z.
Felzer, B.S., T. Cronin, J.M. Reilly, J.M. Melillo, and X. Wang, 2007: Impacts of ozone on trees and crops. Comptes Rendus Geoscience, 339(11–12), 784–798, doi: 10.1016/j.crte.2007.08.008.
Feng, J. et al., 2006: A comparative study of the organic matter in PM2.5 from three Chinese megacities in three different climatic zones. Atmospheric Environment, 40(21), 3983–3994, doi: 10.1016/j.atmosenv.2006.02.017.
Feng, L. et al., 2020: Gridded Emissions for CMIP6. Geoscientific Model Development, 13(2), 461–482, doi: 10.5194/gmd-13-461-2020.
Feng, Z. and K. Kobayashi, 2009: Assessing the impacts of current and future concentrations of surface ozone on crop yield with meta-analysis. Atmospheric Environment, 43(8), 1510–1519, doi: 10.1016/j.atmosenv.2008.11.033.
Ferek, R.J. et al., 2000: Drizzle Suppression in Ship Tracks. Journal of the Atmospheric Sciences, 57(16), 2707–2728, doi: 10.1175/1520-0469(2000)057<2707:dsist>2.0.co;2.
Ferraro, A.J., E.J. Highwood, and A.J. Charlton-Perez, 2011: Stratospheric heating by potential geoengineering aerosols. Geophysical Research Letters, 38(24), L24706, doi: 10.1029/2011gl049761.
Fiebig, M., A. Wiartalla, B. Holderbaum, and S. Kiesow, 2014: Particulate emissions from diesel engines: correlation between engine technology and emissions. Journal of Occupational Medicine and Toxicology, 9(1), 6, doi: 10.1186/1745-6673-9-6.
Finney, D.L., R.M. Doherty, O. Wild, and N.L. Abraham, 2016a: The impact of lightning on tropospheric ozone chemistry using a new global lightning parametrisation. Atmospheric Chemistry and Physics, 16(12), 7507–7522, doi: 10.5194/acp-16-7507-2016.
Finney, D.L., R.M. Doherty, O. Wild, P.J. Young, and A. Butler, 2016b: Response of lightning NOx emissions and ozone production to climate change: Insights from the Atmospheric Chemistry and Climate Model Intercomparison Project. Geophysical Research Letters, 43(10), 5492–5500, doi: 10.1002/2016gl068825.
Finney, D.L. et al., 2018: A projected decrease in lightning under climate change. Nature Climate Change, 8(3), 210–213, doi: 10.1038/s41558-018-0072-6.
Fioletov, V.E. et al., 2016: A global catalogue of large SO2 sources and emissions derived from the Ozone Monitoring Instrument. Atmospheric Chemistry and Physics, 16(18), 11497–11519, doi: 10.5194/acp-16-11497-2016.
Fiore, A.M., V. Naik, and E.M. Leibensperger, 2015: Air Quality and Climate Connections. Journal of the Air & Waste Management Association, 65(6), 645–685, doi: 10.1080/10962247.2015.1040526.
Fiore, A.M. et al., 2012: Global air quality and climate. Chemical Society Reviews, 41(19), 6663–6683, doi: 10.1039/c2cs35095e.
Flechard, C.R. et al., 2020: Carbon–nitrogen interactions in European forests and semi-natural vegetation – Part 1: Fluxes and budgets of carbon, nitrogen and greenhouse gases from ecosystem monitoring and modelling. Biogeosciences, 17(6), 1583–1620, doi: 10.5194/bg-17-1583-2020.
Flemming, J. et al., 2017: The CAMS interim Reanalysis of Carbon Monoxide, Ozone and Aerosol for 2003–2015. Atmospheric Chemistry and Physics, 17(3), 1945–1983, doi: 10.5194/acp-17-1945-2017.
Forestieri, S.D. et al., 2018: Temperature and Composition Dependence of Sea Spray Aerosol Production. Geophysical Research Letters, 45(14), 7218–7225, doi: 10.1029/2018gl078193.
Forster, P.M. et al., 2016: Recommendations for diagnosing effective radiative forcing from climate models for CMIP6. Journal of Geophysical Research: Atmospheres, 121(20), 12460–12475, doi: 10.1002/2016jd025320.
Forster, P.M. et al., 2020: Current and future global climate impacts resulting from COVID-19. Nature Climate Change, 10(10), 913–919, doi: 10.1038/s41558-020-0883-0.
Fortems-Cheiney, A. et al., 2017: A 3°C global RCP8.5 emission trajectory cancels benefits of European emission reductions on air quality. Nature Communications, 8(1), 89, doi: 10.1038/s41467-017-00075-9.
Fowler, D. et al., 2009: Atmospheric composition change: Ecosystems–Atmosphere interactions. Atmospheric Environment, 43(33), 5193–5267, doi: 10.1016/j.atmosenv.2009.07.068.
Fowler, D. et al., 2015: Effects of global change during the 21st century on the nitrogen cycle. Atmospheric Chemistry and Physics, 15(24), 13849–13893, doi: 10.5194/acp-15-13849-2015.
Fowler, D. et al., 2020: A chronology of global air quality. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 378(2183), 20190314, doi: 10.1098/rsta.2019.0314.
Franchin, A. et al., 2018: Airborne and ground-based observations of ammonium-nitrate-dominated aerosols in a shallow boundary layer during intense winter pollution episodes in northern Utah. Atmospheric Chemistry and Physics, 18(23), 17259–17276, doi: 10.5194/acp-18-17259-2018.
Franco, B. et al., 2016: Evaluating ethane and methane emissions associated with the development of oil and natural gas extraction in North America. Environmental Research Letters, 11(4), 044010, doi: 10.1088/1748-9326/11/4/044010.
Freedman, M.A., E.J.E. Ott, and K.E. Marak, 2019: Role of pH in Aerosol Processes and Measurement Challenges. Journal of Physical Chemistry A, 123(7), 1275–1284, doi: 10.1021/acs.jpca.8b10676.
Freney, E. et al., 2018: Aerosol composition and the contribution of SOA formation over Mediterranean forests. Atmospheric Chemistry and Physics, 18(10), 7041–7056, doi: 10.5194/acp-18-7041-2018.
Friedlingstein, P. et al., 2020: Global Carbon Budget 2020. Earth System Science Data, 12(4), 3269–3340, doi: 10.5194/essd-12-3269-2020.
Froyd, K.D. et al., 2009: Aerosol composition of the tropical upper troposphere. Atmospheric Chemistry and Physics, 9(13), 4363–4385, doi: 10.5194/acp-9-4363-2009.
Fu, D. et al., 2019: Direct retrieval of isoprene from satellite-based infrared measurements. Nature Communications, 10(1), 3811, doi: 10.1038/s41467-019-11835-0.
Fu, T.M. and H. Tian, 2019: Climate Change Penalty to Ozone Air Quality: Review of Current Understandings and Knowledge Gaps. Current Pollution Reports, 5(3), 159–171, doi: 10.1007/s40726-019-00115-6.
Fuchs, H. et al., 2013: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation. Nature Geoscience, 6(12), 1023–1026, doi: 10.1038/ngeo1964.
Fuglestvedt, J.S., I.S.A. Isaksen, and W.C. Wang, 1996: Estimates of indirect global warming potentials for CH4, CO and NOx. Climatic Change, 34(3–4), 405–437, doi: 10.1007/bf00139300.
Fuglestvedt, J.S., T. Berntsen, G. Myhre, K. Rypdal, and R.B. Skeie, 2008: Climate forcing from the transport sectors. Proceedings of the National Academy of Sciences, 105(2), 454–458, doi: 10.1073/pnas.0702958104.
Fujimori, S. et al., 2017: SSP3: AIM implementation of Shared Socioeconomic Pathways. Global Environmental Change, 42, 268–283, doi: 10.1016/j.gloenvcha.2016.06.009.
Fuzzi, S. et al., 2007: Overview of the inorganic and organic composition of size-segregated aerosol in Rondônia, Brazil, from the biomass-burning period to the onset of the wet season. Journal of Geophysical Research: Atmospheres, 112(D1), D01201, doi: 10.1029/2005jd006741.
Fyfe, J.C. et al., 2021: Quantifying the influence of short-term emission reductions on climate. Science Advances, 7(10), eabf7133, doi: 10.1126/sciadv.abf7133.
Gabric, A.J., B. Qu, L. Rotstayn, and J.M. Shephard, 2013: Global simulations of the impact on contemporary climate of a perturbation to the sea-to-air flux of dimethylsulfide. Australian Meteorological and Oceanographic Journal, 63(3), 365–376, doi: 10.22499/2.6303.002.
Gallardo, L. et al., 2018: Evolution of air quality in Santiago: The role of mobility and lessons from the science–policy interface. Elementa: Science of the Anthropocene, 6(1), 38, doi: 10.1525/elementa.293.
Galmarini, S. et al., 2017: Technical note: Coordination and harmonization of the multi-scale, multi-model activities HTAP2, AQMEII3, and MICS-Asia3: Simulations, emission inventories, boundary conditions, and model output formats. Atmospheric Chemistry and Physics, 17(2), 1543–1555, doi: 10.5194/acp-17-1543-2017.
Gao, M. et al., 2018: Air quality and climate change, Topic 3 of the Model Inter- Comparison Study for Asia Phase III (MICS-Asia III) – Part 1: Overview and model evaluation. Atmospheric Chemistry and Physics, 18(7), 4859–4884, doi: 10.5194/acp-18-4859-2018.
Garcia-Menendez, F., E. Monier, and N.E. Selin, 2017: The role of natural variability in projections of climate change impacts on U.S. ozone pollution. Geophysical Research Letters, 44(6), 2911–2921, doi: 10.1002/2016gl071565.
Garrido-Perez, J.M., C. Ordóñez, R. García-Herrera, and J.L. Schnell, 2019: The differing impact of air stagnation on summer ozone across Europe. Atmospheric Environment, 219, 117062, doi: 10.1016/j.atmosenv. 2019.117062.
Gasparini, B. and U. Lohmann, 2016: Why cirrus cloud seeding cannot substantially cool the planet. Journal of Geophysical Research: Atmospheres, 121(9), 4877–4893, doi: 10.1002/2015jd024666.
Gasparini, B., Z. McGraw, T. Storelvmo, and U. Lohmann, 2020: To what extent can cirrus cloud seeding counteract global warming?Environmental Research Letters, 15(5), 054002, doi: 10.1088/1748-9326/ab71a3.
Gaubert, B. et al., 2017: Chemical Feedback From Decreasing Carbon Monoxide Emissions. Geophysical Research Letters, 44(19), 9985–9995, doi: 10.1002/2017gl074987.
Gaudel, A. et al., 2018: Tropospheric Ozone Assessment Report: Present-day distribution and trends of tropospheric ozone relevant to climate and global atmospheric chemistry model evaluation. Elementa: Science of the Anthropocene, 6(1), 39, doi: 10.1525/elementa.291.
Gaudel, A. et al., 2020: Aircraft observations since the 1990s reveal increases of tropospheric ozone at multiple locations across the Northern Hemisphere. Science Advances, 6(34), eaba8272, doi: 10.1126/sciadv.aba8272.
Ge, C., C. Zhu, J.S. Francisco, X.C. Zeng, and J. Wang, 2018: A molecular perspective for global modeling of upper atmospheric NH3 from freezing clouds. Proceedings of the National Academy of Sciences, 115(24), 6147–6152, doi: 10.1073/pnas.1719949115.
Geddes, J.A., R. Martin, B.L. Boys, and A. van Donkelaar, 2016: Long-Term Trends Worldwide in Ambient NO2 concentrations Inferred from Satellite Observations. Environmental Health Perspectives, 124(3), 281–289, doi: 10.1289/ehp.1409567.
Genard-Zielinski, A.-C. et al., 2018: Seasonal variations of Quercus pubescens isoprene emissions from an in natura forest under drought stress and sensitivity to future climate change in the Mediterranean area. Biogeosciences, 15(15), 4711–4730, doi: 10.5194/bg-15-4711-2018.
Geng, L. et al., 2014: Nitrogen isotopes in ice core nitrate linked to anthropogenic atmospheric acidity change. Proceedings of the National Academy of Sciences, 111(16), 5808–5812, doi: 10.1073/pnas.1319441111.
Geoffroy, O. et al., 2013: Transient Climate Response in a Two-Layer Energy-Balance Model. Part I: Analytical Solution and Parameter Calibration Using CMIP5 AOGCM Experiments. Journal of Climate, 26(6), 1841–1857, doi: 10.1175/jcli-d-12-00195.1.
Georgoulias, A.K., R.A.J. Van Der, P. Stammes, K. Folkert Boersma, and H.J. Eskes, 2019: Trends and trend reversal detection in 2 decades of tropospheric NO2 satellite observations. Atmospheric Chemistry and Physics, 19(9), 6269–6294, doi: 10.5194/acp-19-6269-2019.
Gettelman, A., R. Lamboll, C.G. Bardeen, P.M. Forster, and D. Watson-Parris, 2021: Climate Impacts of COVID-19 Induced Emission Changes. Geophysical Research Letters, 48(3), e2020GL091805, doi: 10.1029/2020gl091805.
Ghan, S.J., 2013: Technical Note: Estimating aerosol effects on cloud radiative forcing. Atmospheric Chemistry and Physics, 13(19), 9971–9974, doi: 10.5194/acp-13-9971-2013.
Gidden, M.J. et al., 2019: Global emissions pathways under different socioeconomic scenarios for use in CMIP6: A dataset of harmonized emissions trajectories through the end of the century. Geoscientific Model Development, 12(4), 1443–1475, doi: 10.5194/gmd-12-1443-2019.
Ginoux, P., J.M. Prospero, T.E. Gill, N.C. Hsu, and M. Zhao, 2012: Global-scale attribution of anthropogenic and natural dust sources and their emission rates based on MODIS Deep Blue aerosol products. Reviews of Geophysics, 50(3), RG3005, doi: 10.1029/2012rg000388.
Gioda, A., B.S. Amaral, I.L.G. Monteiro, and T.D. Saint’Pierre, 2011: Chemical composition, sources, solubility, and transport of aerosol trace elements in a tropical region. Journal of Environmental Monitoring, 13(8), 2134, doi: 10.1039/c1em10240k.
Gkatzelis, G.I. et al., 2021: The global impacts of COVID-19 lockdowns on urban air quality: a critical review and recommendations. Elementa: Science of the Anthropocene, 9(1), 00176, doi: 10.1525/elementa.2021.00176.
Gliß, J. et al., 2021: AeroCom phase III multi-model evaluation of the aerosol life cycle and optical properties using ground- and space-based remote sensing as well as surface in situ observations. Atmospheric Chemistry and Physics, 21(1), 87–128, doi: 10.5194/acp-21-87-2021.
Gómez-Sanabria, A., L. Höglund-Isaksson, P. Rafaj, and W. Schöpp, 2018: Carbon in global waste and wastewater flows – its potential as energy source under alternative future waste management regimes. Advances in Geosciences, 45, 105–113, doi: 10.5194/adgeo-45-105-2018.
Gonzalez-Abraham, R. et al., 2015: The effects of global change upon United States air quality. Atmospheric Chemistry and Physics, 15(21), 12645–12665, doi: 10.5194/acp-15-12645-2015.
Gratz, L.E., D.A. Jaffe, and J.R. Hee, 2015: Causes of increasing ozone and decreasing carbon monoxide in springtime at the Mt. Bachelor Observatory from 2004 to 2013. Atmospheric Environment, 109, 323–330, doi: 10.1016/j.atmosenv.2014.05.076.
Grewe, V., S. Matthes, and K. Dahlmann, 2019: The contribution of aviation NOx emissions to climate change: are we ignoring methodological flaws?Environmental Research Letters, 14(12), 121003, doi: 10.1088/1748-9326/ab5dd7.
Grewe, V. et al., 2001: Origin and variability of upper tropospheric nitrogen oxides and ozone at northern mid-latitudes. Atmospheric Environment, 35(20), 3421–3433, doi: 10.1016/s1352-2310(01)00134-0.
Griffiths, P.T. et al., 2021: Tropospheric ozone in CMIP6 Simulations. Atmospheric Chemistry and Physics, 21(5), 4187–4218, doi: 10.5194/acp-21-4187-2021.
Grythe, H., J. Ström, R. Krejci, P. Quinn, and A. Stohl, 2014: A review of sea-spray aerosol source functions using a large global set of sea salt aerosol concentration measurements. Atmospheric Chemistry and Physics, 14(3), 1277–1297, doi: 10.5194/acp-14-1277-2014.
Gschrey, B., W. Schwarz, C. Elsner, and R. Engelhardt, 2011: High increase of global F-gas emissions until 2050. Greenhouse Gas Measurement and Management, 1(2), 85–92, doi: 10.1080/20430779.2011.579352.
Guenther, A.B. et al., 2012: The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): An extended and updated framework for modeling biogenic emissions. Geoscientific Model Development, 5(6), 1471–1492, doi: 10.5194/gmd-5-1471-2012.
Guo, H. et al., 2016: Fine particle pH and the partitioning of nitric acid during winter in the northeastern United States. Journal of Geophysical Research: Atmospheres, 121(17), 10355–10376, doi: 10.1002/2016jd025311.
Guo, H. et al., 2018: Effectiveness of ammonia reduction on control of fine particle nitrate. Atmospheric Chemistry and Physics, 18(16), 12241–12256, doi: 10.5194/acp-18-12241-2018.
Guo, S. et al., 2014: Elucidating severe urban haze formation in China. Proceedings of the National Academy of Sciences, 111(49), 17373–17378, doi: 10.1073/pnas.1419604111.
Guttikunda, S.K., R. Goel, and P. Pant, 2014: Nature of air pollution, emission sources, and management in the Indian cities. Atmospheric Environment, 95, 501–510, doi: 10.1016/j.atmosenv.2014.07.006.
Haines, A. et al., 2017: Short-lived climate pollutant mitigation and the Sustainable Development Goals. Nature Climate Change, 7(12), 863–869, doi: 10.1038/s41558-017-0012-x.
Hammer, M.S. et al., 2018: Insight into global trends in aerosol composition from 2005 to 2015 inferred from the OMI Ultraviolet Aerosol Index. Atmospheric Chemistry and Physics, 18(11), 8097–8112, doi: 10.5194/acp-18-8097-2018.
Hand, J.L., K.A. Gebhart, B.A. Schichtel, and W.C. Malm, 2012: Increasing trends in wintertime particulate sulfate and nitrate ion concentrations in the Great Plains of the United States (2000–2010). Atmospheric Environment, 55, 107–110, doi: 10.1016/j.atmosenv.2012.03.050.
Hand, J.L., B.A. Schichtel, W.C. Malm, and N.H. Frank, 2013: Spatial and Temporal Trends in PM2.5 organic and Elemental Carbon across the United States. Advances in Meteorology, 2013, 1–13, doi: 10.1155/2013/367674.
Hantson, S., W. Knorr, G. Schurgers, T.A.M. Pugh, and A. Arneth, 2017: Global isoprene and monoterpene emissions under changing climate, vegetation, CO2 and land use. Atmospheric Environment, 155, 35–45, doi: 10.1016/j.atmosenv.2017.02.010.
Harmsen, M.J.H.M. et al., 2015: How well do integrated assessment models represent non-CO2 radiative forcing?Climatic Change, 133(4), 565–582, doi: 10.1007/s10584-015-1485-0.
Harmsen, M.J.H.M. et al., 2020a: Taking some heat off the NDCs? The limited potential of additional short-lived climate forcers’ mitigation. Climatic Change, 163(3), 1443–1461, doi: 10.1007/s10584-019-02436-3.
Harmsen, M.J.H.M. et al., 2020b: Co-benefits of black carbon mitigation for climate and air quality. Climatic Change, 163(3), 1519–1538, doi: 10.1007/s10584-020-02800-8.
Hartmann, D.L. et al., 2013: Observations: Atmosphere and Surface. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change[Stocker, T.F., D. Qin, G.-K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P.M. Midgley (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 159–254, doi: 10.1017/cbo9781107415324.008.
Hastings, M.G., J.C. Jarvis, and E.J. Steig, 2009: Anthropogenic Impacts on Nitrogen Isotopes of Ice-Core Nitrate. Science, 324(5932), 1288, doi: 10.1126/science.1170510.
Hauglustaine, D.A., Y. Balkanski, and M. Schulz, 2014: A global model simulation of present and future nitrate aerosols and their direct radiative forcing of climate. Atmospheric Chemistry and Physics, 14(20), 11031–11063, doi: 10.5194/acp-14-11031-2014.
He, H., X.Z. Liang, and D.J. Wuebbles, 2018: Effects of emissions change, climate change and long-range transport on regional modeling of future U.S. particulate matter pollution and speciation. Atmospheric Environment, 179, 166–176, doi: 10.1016/j.atmosenv.2018.02.020.
Heald, C.L. and D. Spracklen, 2015: Land Use Change Impacts on Air Quality and Climate. Chemical Reviews, 115(10), 4476–4496, doi: 10.1021/cr500446g.
Heald, C.L. and J.A. Geddes, 2016: The impact of historical land use change from 1850 to 2000 on secondary particulate matter and ozone. Atmospheric Chemistry and Physics, 16(23), 14997–15010, doi: 10.5194/acp-16-14997-2016.
Heald, C.L. et al., 2014: Contrasting the direct radiative effect and direct radiative forcing of aerosols. Atmospheric Chemistry and Physics, 14(11), 5513–5527, doi: 10.5194/acp-14-5513-2014.
Heinze, C. et al., 2019: ESD Reviews: Climate feedbacks in the Earth system and prospects for their evaluation. Earth System Dynamics, 10(3), 379–452, doi: 10.5194/esd-10-379-2019.
Hess, P., D. Kinnison, and Q. Tang, 2015: Ensemble simulations of the role of the stratosphere in the attribution of northern extratropical tropospheric ozone variability. Atmospheric Chemistry and Physics, 15(5), 2341–2365, doi: 10.5194/acp-15-2341-2015.
Hock, R. et al., 2019: High Mountain Areas. In: IPCC Special Report on the Ocean and Cryosphere in a Changing Climate[Pörtner, H.-O., D.C. Roberts, V. Masson-Delmotte, P. Zhai, M. Tignor, E. Poloczanska, K. Mintenbeck, A. Alegría, M. Nicolai, A. Okem, J. Petzold, B. Rama, and N.M. Weyer (eds.)]. In Press, pp. 131–202, www.ipcc.ch/srocc/chapter/chapter-2.
Hodgson, A.K. et al., 2018: Near-field emission profiling of tropical forest and Cerrado fires in Brazil during SAMBBA 2012. Atmospheric Chemistry and Physics, 18(8), 5619–5638, doi: 10.5194/acp-18-5619-2018.
Hodzic, A. et al., 2016: Rethinking the global secondary organic aerosol (SOA) budget: stronger production, faster removal, shorter lifetime. Atmospheric Chemistry and Physics, 16(12), 7917–7941, doi: 10.5194/acp-16-7917-2016.
Hodzic, A. et al., 2020: Characterization of organic aerosol across the global remote troposphere: a comparison of ATom measurements and global chemistry models. Atmospheric Chemistry and Physics, 20(8), 4607–4635, doi: 10.5194/acp-20-4607-2020.
Hoesly, R. et al., 2019: Community Emissions Data System (Version Dec-23-2019). Zenodo. Retrieved from: https://doi.org/10.5281/zenodo.3592073.
Hoesly, R.M. et al., 2018: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS). Geoscientific Model Development, 11(1), 369–408, doi: 10.5194/gmd-11-369-2018.
Hofzumahaus, A. et al., 2009: Amplified Trace Gas Removal in the Troposphere. Science, 324(5935), 1702–1704, doi: 10.1126/science.1164566.
Höglund-Isaksson, L., 2012: Global anthropogenic methane emissions 2005–2030: technical mitigation potentials and costs. Atmospheric Chemistry and Physics, 12(19), 9079–9096, doi: 10.5194/acp-12-9079-2012.
Höglund-Isaksson, L., A. Gómez-Sanabria, Z. Klimont, P. Rafaj, and W. Schöpp, 2020: Technical potentials and costs for reducing global anthropogenic methane emissions in the 2050 timeframe – results from the GAINS model. Environmental Research Communications, 2(2), 025004, doi: 10.1088/2515-7620/ab7457.
Höglund-Isaksson, L. et al., 2017: Cost estimates of the Kigali Amendment to phase-down hydrofluorocarbons. Environmental Science & Policy, 75, 138–147, doi: 10.1016/j.envsci.2017.05.006.
Hollaway, M.J., S.R. Arnold, W.J. Collins, G. Folberth, and A. Rap, 2017: Sensitivity of midnineteenth century tropospheric ozone to atmospheric chemistry-vegetation interactions. Journal of Geophysical Research: Atmospheres, 122(4), 2452–2473, doi: 10.1002/2016jd025462.
Holmes, C.D., 2018: Methane Feedback on Atmospheric Chemistry: Methods, Models, and Mechanisms. Journal of Advances in Modeling Earth Systems, 10(4), 1087–1099, doi: 10.1002/2017ms001196.
Holmes, C.D., M.J. Prather, O.A. Søvde, and G. Myhre, 2013: Future methane, hydroxyl, and their uncertainties: Key climate and emission parameters for future predictions. Atmospheric Chemistry and Physics, 13(1), 285–302, doi: 10.5194/acp-13-285-2013.
Hong, C. et al., 2019: Impacts of climate change on future air quality and human health in China. Proceedings of the National Academy of Sciences, 116(35), 17193–17200, doi: 10.1073/pnas.1812881116.
Hopcroft, P.O., P.J. Valdes, F.M. O’Connor, J.O. Kaplan, and D.J. Beerling, 2017: Understanding the glacial methane cycle. Nature Communications, 8, 14383, doi: 10.1038/ncomms14383.
Höpfner, M. et al., 2016: First detection of ammonia (NH3) in the Asian summer monsoon upper troposphere. Atmospheric Chemistry and Physics, 16(22), 14357–14369, doi: 10.5194/acp-16-14357-2016.
Höpfner, M. et al., 2019: Ammonium nitrate particles formed in upper troposphere from ground ammonia sources during Asian monsoons. Nature Geoscience, 12(8), 608–612, doi: 10.1038/s41561-019-0385-8.
Horowitz, L.W. et al., 2020: The GFDL Global Atmospheric Chemistry–Climate Model AM4.1: Model Description and Simulation Characteristics. Journal of Advances in Modeling Earth Systems, 12(10), e2019MS002032, doi: 10.1029/2019ms002032.
Hoshika, Y. et al., 2015: Ozone-induced stomatal sluggishness changes carbon and water balance of temperate deciduous forests. Scientific Reports, 5(1), 9871, doi: 10.1038/srep09871.
Hossaini, R. et al., 2015: Efficiency of short-lived halogens at influencing climate through depletion of stratospheric ozone. Nature Geoscience, 8(3), 186–190, doi: 10.1038/ngeo2363.
Hossaini, R. et al., 2017: The increasing threat to stratospheric ozone from dichloromethane. Nature Communications, 8, 15962, doi: 10.1038/ncomms15962.
Hou, P. and S. Wu, 2016: Long-term Changes in Extreme Air Pollution Meteorology and the Implications for Air Quality. Scientific Reports, 6(1), 23792, doi: 10.1038/srep23792.
Hou, P., S. Wu, J.L. McCarty, and Y. Gao, 2018: Sensitivity of atmospheric aerosol scavenging to precipitation intensity and frequency in the context of global climate change. Atmospheric Chemistry and Physics, 18(11), 8173–8182, doi: 10.5194/acp-18-8173-2018.
Huang, K. and J.S. Fu, 2016: A global gas flaring black carbon emission rate dataset from 1994 to 2012. Scientific Data, 3(1), 160104, doi: 10.1038/sdata.2016.104.
Huang, K. et al., 2015: Russian anthropogenic black carbon: Emission reconstruction and Arctic black carbon simulation. Journal of Geophysical Research: Atmospheres, 120(21), 11306–11333, doi: 10.1002/2015jd023358.
Huang, R.-J. et al., 2014: High secondary aerosol contribution to particulate pollution during haze events in China. Nature, 514(7521), 218–222, doi: 10.1038/nature13774.
Huang, X. et al., 2021: Enhanced secondary pollution offset reduction of primary emissions during COVID-19 lockdown in China. National Science Review, 8(2), nwaa137, doi: 10.1093/nsr/nwaa137.
Huang, Y., S. Wu, M.K. Dubey, and N.H.F. French, 2013: Impact of aging mechanism on model simulated carbonaceous aerosols. Atmospheric Chemistry and Physics, 13(13), 6329–6343, doi: 10.5194/acp-13-6329-2013.
Huang, Y., N. Unger, K. Harper, and C. Heyes, 2020: Global Climate and Human Health Effects of the Gasoline and Diesel Vehicle Fleets. GeoHealth, 4(3), e2019GH000240, doi: 10.1029/2019gh000240.
Huang, Y. et al., 2018: Global radiative effects of solid fuel cookstove aerosol emissions. Atmospheric Chemistry and Physics, 18(8), 5219–5233, doi: 10.5194/acp-18-5219-2018.
Hudman, R.C. et al., 2012: Steps towards a mechanistic model of global soil nitric oxide emissions: implementation and space based-constraints. Atmospheric Chemistry and Physics, 12(16), 7779–7795, doi: 10.5194/acp-12-7779-2012.
Iizuka, Y. et al., 2018: A 60 Year Record of Atmospheric Aerosol Depositions Preserved in a High-Accumulation Dome Ice Core, Southeast Greenland. Journal of Geophysical Research: Atmospheres, 123(1), 574–589, doi: 10.1002/2017jd026733.
Im, U. et al., 2012: Summertime aerosol chemical composition in the Eastern Mediterranean and its sensitivity to temperature. Atmospheric Environment, 50, 164–173, doi: 10.1016/j.atmosenv.2011.12.044.
IMO, 2016: Effective Date of Implementation of the Fuel Oil Standard in Regulation 14.1.3 of MARPOL Annex VI. Annex 6. Resolution MEPC. 280(70) (Adopted on 28 October 2016). In: MARPOL Annex VI. MEPC 70/18/Add.1, International Maritime Organization (IMO), London, UK, https://wwwcdn.imo.org/localresources/en/OurWork/Environment/Documents/280(70).pdf .
Inness, A. et al., 2019: The CAMS reanalysis of atmospheric composition. Atmospheric Chemistry and Physics, 19(6), 3515–3556, doi: 10.5194/acp-19-3515-2019.
IPCC, 2019a: Climate Change and Land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems [Shukla, P.R., J. Skea, E.C. Buendia, V. Masson-Delmotte, H.-O. Pörtner, D.C. Roberts, P. Zhai, R. Slade, S. Connors, R. Diemen, M. Ferrat, E. Haughey, S. Luz, S. Neogi, M. Pathak, J. Petzold, J.P. Pereira, P. Vyas, E. Huntley, K. Kissick, M. Belkacemi, and J. Malley (eds.)]. In Press, 896 pp., www.ipcc.ch/srccl.
IPCC, 2019b: IPCC Special Report on the Ocean and Cryosphere in a Changing Climate [Pörtner, H.-O., D.C. Roberts, V. Masson-Delmotte, P. Zhai, M. Tignor, E. Poloczanska, K. Mintenbeck, A. Alegría, M. Nicolai, A. Okem, J. Petzold, B. Rama, and N.M. Weyer (eds.)]. In Press, 755 pp., www.ipcc.ch/report/srocc/.
Irie, H., T. Muto, S. Itahashi, J.-I. Kurokawa, and I. Uno, 2016: Turnaround of Tropospheric Nitrogen Dioxide Pollution Trends in China, Japan, and South Korea. SOLA, 12, 170–174, doi: 10.2151/sola.2016-035.
Jackson, L.S., J.A. Crook, and P.M. Forster, 2016: An intensified hydrological cycle in the simulation of geoengineering by cirrus cloud thinning using ice crystal fall speed changes. Journal of Geophysical Research: Atmospheres, 121(12), 6822–6840, doi: 10.1002/2015jd024304.
Jakob, M. and J.C. Steckel, 2016: Implications of climate change mitigation for sustainable development. Environmental Research Letters, 11(10), 104010, doi: 10.1088/1748-9326/11/10/104010.
Janssens-Maenhout, G. et al., 2019: EDGAR v4.3.2 Global (Atlas of the three major greenhouse gas emissions for the period 1970–2012. Earth System Science Data, 11(3), 959–1002, doi: 10.5194/essd-11-959-2019.
Jardine, K.J. et al., 2016: Monoterpene ‘thermometer’ of tropical forest-atmosphere response to climate warming. Plant, Cell & Environment, 40(3), 441–452, doi: 10.1111/pce.12879.
Jia, G. et al., 2019: Land–climate interactions. In: Climate Change and Land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems[Shukla, P.R., J. Skea, E.C. Buendia, V. Masson-Delmotte, H.-O. Pörtner, D.C. Roberts, P. Zhai, R. Slade, S. Connors, R. Diemen, M. Ferrat, E. Haughey, S. Luz, S. Neogi, M. Pathak, J. Petzold, J. Portugal Pereira, E.H. P. Vyas, K. Kissick, M. Belkacemi, and J. Malley (eds.)]. In Press, pp. 131–247, www.ipcc.ch/srccl/chapter/chapter-2.
Jiang, H. et al., 2013: Projected effect of 2000–2050 changes in climate and emissions on aerosol levels in China and associated transboundary transport. Atmospheric Chemistry and Physics, 13(16), 7937–7960, doi: 10.5194/acp-13-7937-2013.
Jiang, L. and B.C. O’Neill, 2017: Global urbanization projections for the Shared Socioeconomic Pathways. Global Environmental Change, 42, 193–199, doi: 10.1016/j.gloenvcha.2015.03.008.
Jiang, Z. et al., 2017: A 15-year record of CO emissions constrained by MOPITT CO observations. Atmospheric Chemistry and Physics, 17(7), 4565–4583, doi: 10.5194/acp-17-4565-2017.
Jiang, Z. et al., 2018: Unexpected slowdown of US pollutant emission reduction in the past decade. Proceedings of the National Academy of Sciences, 115(20), 5099–5104, doi: 10.1073/pnas.1801191115.
Jiao, C. et al., 2014: An AeroCom assessment of black carbon in Arctic snow and sea ice. Atmospheric Chemistry and Physics, 14(5), 2399–2417, doi: 10.5194/acp-14-2399-2014.
Jing, P., Z. Lu, and A.L. Steiner, 2017: The ozone–climate penalty in the Midwestern U.S. Atmospheric Environment, 170, 130–142, doi: 10.1016/j.atmosenv.2017.09.038.
Jo, D.S., R.J. Park, S. Lee, S.W. Kim, and X. Zhang, 2016: A global simulation of brown carbon: Implications for photochemistry and direct radiative effect. Atmospheric Chemistry and Physics, 16(5), 3413–3432, doi: 10.5194/acp-16-3413-2016.
John, J.G., A.M. Fiore, V. Naik, L.W. Horowitz, and J.P. Dunne, 2012: Climate versus emission drivers of methane lifetime against loss by tropospheric OH from 1860–2100. Atmospheric Chemistry and Physics, 12(24), 12021–12036, doi: 10.5194/acp-12-12021-2012.
Jones, A. and J.M. Haywood, 2012: Sea-spray geoengineering in the HadGEM2-ES earth-system model: Radiative impact and climate response. Atmospheric Chemistry and Physics, doi: 10.5194/acp-12-10887-2012.
Jones, A., J. Haywood, and O. Boucher, 2011: A comparison of the climate impacts of geoengineering by stratospheric SO2 injection and by brightening of marine stratocumulus cloud. Atmospheric Science Letters, 12(2), 176–183, doi: 10.1002/asl.291.
Jones, A.C., J.M. Haywood, and A. Jones, 2016: Climatic impacts of stratospheric geoengineering with sulfate, black carbon and titania injection. Atmospheric Chemistry and Physics, 16(5), 2843–2862, doi: 10.5194/acp-16-2843-2016.
Jones, C.D., 2003: Strong carbon cycle feedbacks in a climate model with interactive CO2and sulphate aerosols. Geophysical Research Letters, 30(9), 1479, doi: 10.1029/2003gl016867.
Jonson, J.E., M. Gauss, M. Schulz, J.-P. Jalkanen, and H. Fagerli, 2020: Effects of global ship emissions on European air pollution levels. Atmospheric Chemistry and Physics, 20(19), 11399–11422, doi: 10.5194/acp-20-11399-2020.
Jonson, J.E. et al., 2017: Impact of excess NOx emissions from diesel cars on air quality, public health and eutrophication in Europe. Environmental Research Letters, 12(9), 94017, doi: 10.1088/1748-9326/aa8850.
Kaltsonoudis, C., E. Kostenidou, K. Florou, M. Psichoudaki, and S.N. Pandis, 2016: Temporal variability and sources of VOCs in urban areas of the eastern Mediterranean. Atmospheric Chemistry and Physics, 16(23), 14825–14842, doi: 10.5194/acp-16-14825-2016.
Kanakidou, M., S. Myriokefalitakis, and K. Tsigaridis, 2018: Aerosols in atmospheric chemistry and biogeochemical cycles of nutrients. Environmental Research Letters, 13(6), 063004, doi: 10.1088/1748-9326/aabcdb.
Kanaya, Y. et al., 2020: Rapid reduction in black carbon emissions from China: evidence from 2009–2019 observations on Fukue Island, Japan. Atmospheric Chemistry and Physics, 20(11), 6339–6356, doi: 10.5194/acp-20-6339-2020.
Kang, S. et al., 2002: Twentieth century increase of atmospheric ammonia recorded in Mount Everest ice core. Journal of Geophysical Research: Atmospheres, 107(D21), ACL 13-1-ACL 13-9, doi: 10.1029/2001jd001413.
Karset, I.H.H. et al., 2018: Strong impacts on aerosol indirect effects from historical oxidant changes. Atmospheric Chemistry and Physics, 18(10), 7669–7690, doi: 10.5194/acp-18-7669-2018.
Kasoar, M., D. Shawki, and A. Voulgarakis, 2018: Similar spatial patterns of global climate response to aerosols from different regions. npj Climate and Atmospheric Science, 1(1), 12, doi: 10.1038/s41612-018-0022-z.
Kasoar, M. et al., 2016: Regional and global temperature response to anthropogenic SO2 emissions from China in three climate models. Atmospheric Chemistry and Physics, 16(15), 9785–9804, doi: 10.5194/acp-16-9785-2016.
Keeble, J. et al., 2021: Evaluating stratospheric ozone and water vapour changes in CMIP6 models from 1850 to 2100. Atmospheric Chemistry and Physics, 21(6), 5015–5061, doi: 10.5194/acp-21-5015-2021.
Kekonen, T. et al., 2005: The 800 year long ion record from the Lomonosovfonna (Svalbard) ice core. Journal of Geophysical Research: Atmospheres, 110(D7), D07304, doi: 10.1029/2004jd005223.
Kerr, G.H. and D.W. Waugh, 2018: Connections between summer air pollution and stagnation. Environmental Research Letters, 13(8), 084001, doi: 10.1088/1748-9326/aad2e2.
Kholod, N., M. Evans, and T. Kuklinski, 2016: Russia’s black carbon emissions: Focus on diesel sources. Atmospheric Chemistry and Physics, 16(17), 11267–11281, doi: 10.5194/acp-16-11267-2016.
Kirkby, J. et al., 2011: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature, 476(7361), 429–433, doi: 10.1038/nature10343.
Kirkevåg, A. et al., 2018: A production-tagged aerosol module for Earth system models, OsloAero5.3 – extensions and updates for CAM5.3-Oslo. Geoscientific Model Development, 11(10), 3945–3982, doi: 10.5194/gmd-11-3945-2018.
Kirtman, B. et al., 2013: Near-term Climate Change: Projections and Predictability. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change[Stocker, T.F., D. Qin, G.-K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P.M. Midgley (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, USA, pp. 953–1028, doi: 10.1017/cbo9781107415324.023.
Kleinschmitt, C., O. Boucher, and U. Platt, 2018: Sensitivity of the radiative forcing by stratospheric sulfur geoengineering to the amount and strategy of the SO2 injection studied with the LMDZ-S3A model. Atmospheric Chemistry and Physics, 18(4), 2769–2786, doi: 10.5194/acp-18-2769-2018.
Klimont, Z. and W. Winiwarter, 2015: Estimating Costs and Potential for Reduction of Ammonia Emissions from Agriculture in the GAINS Model. In: Costs of Ammonia Abatement and the ClimateCo-Benefits[Reis, S., C. Howard, and M.A. Sutton (eds.)]. Springer, Dordrecht, The Netherlands, pp. 233–261, doi: 10.1007/978-94-017-9722-1_9.
Klimont, Z. et al., 2017a: Global anthropogenic emissions of particulate matter including black carbon. Atmospheric Chemistry and Physics, 17(14), 8681–8723, doi: 10.5194/acp-17-8681-2017.
Klimont, Z. et al., 2017b: Bridging the gap – The role of short-lived climate pollutants. In: The Emissions Gap Report 2017. United Nations Environment Programme (UNEP), Nairobi, Kenya, pp. 48–57, www.unep.org/resources/emissions-gap-report-2017.
Kloster, S. et al., 2007: Response of dimethylsulfide (DMS) in the ocean and atmosphere to global warming. Journal of Geophysical Research: Biogeosciences, 112(G3), G03005, doi: 10.1029/2006jg000224.
Kodros, J.K. et al., 2015: Uncertainties in global aerosols and climate effects due to biofuel emissions. Atmospheric Chemistry and Physics, 15(15), 8577–8596, doi: 10.5194/acp-15-8577-2015.
Kok, J.F., E.J.R. Parteli, T.I. Michaels, and D.B. Karam, 2012: The physics of wind-blown sand and dust. Reports on Progress in Physics, 75(10), 106901, doi: 10.1088/0034-4885/75/10/106901.
Kok, J.F., D.S. Ward, N.M. Mahowald, and A.T. Evan, 2018: Global and regional importance of the direct dust–climate feedback. Nature Communications, 9(1), 241, doi: 10.1038/s41467-017-02620-y.
Kok, J.F. et al., 2014: An improved dust emission model – Part 1: Model description and comparison against measurements. Atmospheric Chemistry and Physics, 14(23), 13023–13041, doi: 10.5194/acp-14-13023-2014.
Kok, J.F. et al., 2017: Smaller desert dust cooling effect estimated from analysis of dust size and abundance. Nature Geoscience, 10(4), 274–278, doi: 10.1038/ngeo2912.
Koshak, W., H. Peterson, A. Biazar, M. Khan, and L. Wang, 2014: The NASA Lightning Nitrogen Oxides Model (LNOM): Application to air quality modeling. Atmospheric Research, 135–136, 363–369, doi: 10.1016/j.atmosres.2012.12.015.
Kremser, S. et al., 2016: Stratospheric aerosol – Observations, processes, and impact on climate. Reviews of Geophysics, 54(2), 278–335, doi: 10.1002/2015rg000511.
Kristiansen, N.I. et al., 2016: Evaluation of observed and modelled aerosol lifetimes using radioactive tracers of opportunity and an ensemble of 19 global models. Atmospheric Chemistry and Physics, 16(5), 3525–3561, doi: 10.5194/acp-16-3525-2016.
Kristjánsson, J.E., H. Muri, and H. Schmidt, 2015: The hydrological cycle response to cirrus cloud thinning. Geophysical Research Letters, 42(24), 10807–10815, doi: 10.1002/2015gl066795.
Kroll, J.H. et al., 2020: The complex chemical effects of COVID-19 shutdowns on air quality. Nature Chemistry, 12(9), 777–779, doi: 10.1038/s41557-020-0535-z.
Krotkov, N.A. et al., 2016: Aura OMI observations of regional SO2 and NO2 pollution changes from 2005 to 2015. Atmospheric Chemistry and Physics, 16(7), 4605–4629, doi: 10.5194/acp-16-4605-2016.
Krzyzanowski, M. and A. Cohen, 2008: Update of WHO air quality guidelines. Air Quality, Atmosphere & Health, 1(1), 7–13, doi: 10.1007/s11869-008-0008-9.
Kulmala, M. et al., 2004: A new feedback mechanism linking forests, aerosols, and climate. Atmospheric Chemistry and Physics, 4(2), 557–562, doi: 10.5194/acp-4-557-2004.
Kumar, A. et al., 2013: Free-troposphere ozone and carbon monoxide over the North Atlantic for 2001–2011. Atmospheric Chemistry and Physics, 13(24), 12537–12547, doi: 10.5194/acp-13-12537-2013.
Kuprov, R. et al., 2014: Composition and secondary formation of fine particulate matter in the Salt Lake Valley: Winter 2009. Journal of the Air & Waste Management Association, 64(8), 957–969, doi: 10.1080/10962247.2014.903878.
Kutzner, R.D. et al., 2018: Long-term monitoring of black carbon across Germany. Atmospheric Environment, 185, 41–52, doi: 10.1016/j.atmosenv.2018.04.039.
Kuzu, S.L. et al., 2020: Black carbon and size-segregated elemental carbon, organic carbon compositions in a megacity: a case study for Istanbul. Air Quality, Atmosphere & Health, 13(7), 827–837, doi: 10.1007/s11869-020-00839-1.
Laakso, A., H. Korhonen, S. Romakkaniemi, and H. Kokkola, 2017: Radiative and climate effects of stratospheric sulfur geoengineering using seasonally varying injection areas. Atmospheric Chemistry and Physics, 17(11), 6957–6974, doi: 10.5194/acp-17-6957-2017.
Lacressonnière, G. et al., 2016: Impacts of regional climate change on air quality projections and associated uncertainties. Climatic Change, 136(2), 309–324, doi: 10.1007/s10584-016-1619-z.
Lacressonnière, G. et al., 2017: Particulate matter air pollution in Europe in a +2°C warming world. Atmospheric Environment, 154, 129–140, doi: 10.1016/j.atmosenv.2017.01.037.
Laj, P. et al., 2020: A global analysis of climate-relevant aerosol properties retrieved from the network of Global Atmosphere Watch (GAW) near-surface observatories. Atmospheric Measurement Techniques, 13(8), 4353–4392, doi: 10.5194/amt-13-4353-2020.
Lamarque, J.-F. et al., 2010: Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: methodology and application. Atmospheric Chemistry and Physics, 10(15), 7017–7039, doi: 10.5194/acp-10-7017-2010.
Lamarque, J.-F. et al., 2013a: Multi-model mean nitrogen and sulfur deposition from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP): evaluation of historical and projected future changes. Atmospheric Chemistry and Physics, 13(16), 7997–8018, doi: 10.5194/acp-13-7997-2013.
Lamarque, J.-F. et al., 2013b: The Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP): overview and description of models, simulations and climate diagnostics. Geoscientific Model Development, 6(1), 179–206, doi: 10.5194/gmd-6-179-2013.
Lamsal, L.N. et al., 2015: U.S. NO2 trends (2005–2013): EPA Air Quality System (AQS) data versus improved observations from the Ozone Monitoring Instrument (OMI). Atmospheric Environment, 110, 130–143, doi: 10.1016/j.atmosenv.2015.03.055.
Lana, A. et al., 2011: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean. Global Biogeochemical Cycles, 25(1), GB1004, doi: 10.1029/2010gb003850.
Landry, J.-S., H.D. Matthews, and N. Ramankutty, 2015: A global assessment of the carbon cycle and temperature responses to major changes in future fire regime. Climatic Change, 133(2), 179–192, doi: 10.1007/s10584-015-1461-8.
Languille, B. et al., 2020: Wood burning: A major source of Volatile Organic Compounds during wintertime in the Paris region. Science of The Total Environment, 711, 135055, doi: 10.1016/j.scitotenv.2019.135055.
Latham, J. et al., 2008: Global temperature stabilization via controlled albedo enhancement of low-level maritime clouds. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 366(1882), 3969–3987, doi: 10.1098/rsta.2008.0137.
Lathière, J., C.N. Hewitt, and D.J. Beerling, 2010: Sensitivity of isoprene emissions from the terrestrial biosphere to 20th century changes in atmospheric CO2 concentration, climate, and land use. Global Biogeochemical Cycles, 24(1), GB1004, doi: 10.1029/2009gb003548.
Lauer, A., V. Eyring, J. Hendricks, P. Jöckel, and U. Lohmann, 2007: Global model simulations of the impact of ocean-going ships on aerosols, clouds, and the radiation budget. Atmospheric Chemistry and Physics, 7(19), 5061–5079, doi: 10.5194/acp-7-5061-2007.
Lauwaet, D. et al., 2014: The effect of climate change and emission scenarios on ozone concentrations over Belgium: a high-resolution model study for policy support. Atmospheric Chemistry and Physics, 14(12), 5893–5904, doi: 10.5194/acp-14-5893-2014.
Lawrence, M.G. et al., 2018: Evaluating climate geoengineering proposals in the context of the Paris Agreement temperature goals. Nature Communications, 9(1), 3734, doi: 10.1038/s41467-018-05938-3.
Le, T. et al., 2020: Unexpected air pollution with marked emission reductions during the COVID-19 outbreak in China. Science, 369(6504), 702–706, doi: 10.1126/science.abb7431.
Le Quéré, C. et al., 2020: Temporary reduction in daily global CO2 emissions during the COVID-19 forced confinement. Nature Climate Change, 10(7), 647–653, doi: 10.1038/s41558-020-0797-x.
Lee, D.S. et al., 2021: The contribution of global aviation to anthropogenic climate forcing for 2000 to 2018. Atmospheric Environment, 244, 117834, doi: 10.1016/j.atmosenv.2020.117834.
Lee, Y., D.T. Shindell, G. Faluvegi, and R.W. Pinder, 2016: Potential impact of a US climate policy and air quality regulations on future air quality and climate change. Atmospheric Chemistry and Physics, 16(8), 5323–5342, doi: 10.5194/acp-16-5323-2016.
Lee, Y.H. et al., 2013: Evaluation of preindustrial to present-day black carbon and its albedo forcing from Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmospheric Chemistry and Physics, 13(5), 2607–2634, doi: 10.5194/acp-13-2607-2013.
Lehtipalo, K. et al., 2018: Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors. Science Advances, 4(12), eaau5363, doi: 10.1126/sciadv.aau5363.
Lelieveld, J., 2017: Clean air in the Anthropocene. Faraday Discussions, 200, 693–703, doi: 10.1039/c7fd90032e.
Lelieveld, J., S. Gromov, A. Pozzer, and D. Taraborrelli, 2016: Global tropospheric hydroxyl distribution, budget and reactivity. Atmospheric Chemistry and Physics, 16(19), 12477–12493, doi: 10.5194/acp-16-12477-2016.
Lelieveld, J., S. Beirle, C. Hörmann, G. Stenchikov, and T. Wagner, 2015a: Abrupt recent trend changes in atmospheric nitrogen dioxide over the Middle East. Science Advances, 1(7), e1500498, doi: 10.1126/sciadv.1500498.
Lelieveld, J., J.S. Evans, M. Fnais, D. Giannadaki, and A. Pozzer, 2015b: The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature, 525(7569), 367–371, doi: 10.1038/nature15371.
Lelieveld, J. et al., 2014: Model projected heat extremes and air pollution in the eastern Mediterranean and Middle East in the twenty-first century. Regional Environmental Change, 14(5), 1937–1949, doi: 10.1007/s10113-013-0444-4.
Lelieveld, J. et al., 2019: Effects of fossil fuel and total anthropogenic emission removal on public health and climate. Proceedings of the National Academy of Sciences, 116(15), 7192–7197, doi: 10.1073/pnas.1819989116.
Lemaire, V.E.P., A. Colette, and L. Menut, 2016: Using statistical models to explore ensemble uncertainty in climate impact studies: The example of air pollution in Europe. Atmospheric Chemistry and Physics, 16(4), 2559–2574, doi: 10.5194/acp-16-2559-2016.
Lewinschal, A. et al., 2019: Local and remote temperature response of regional SO2 emissions. Atmospheric Chemistry and Physics, 19(4), 2385–2403, doi: 10.5194/acp-19-2385-2019.
Li, H. et al., 2016: Size-Dependent Characterization of Atmospheric Particles during Winter in Beijing. Atmosphere, 7(3), 36, doi: 10.3390/atmos7030036.
Li, K. et al., 2019: Anthropogenic drivers of 2013–2017 trends in summer surface ozone in China. Proceedings of the National Academy of Sciences, 116(2), 422–427, doi: 10.1073/pnas.1812168116.
Li, K. et al., 2020: Increases in surface ozone pollution in China from 2013 to 2019: anthropogenic and meteorological influences. Atmospheric Chemistry and Physics, 20(19), 11423–11433, doi: 10.5194/acp-20-11423-2020.
Li, L. et al., 2010: Composition, source, mass closure of PM2.5 aerosols for four forests in eastern China. Journal of Environmental Sciences, 22(3), 405–412, doi: 10.1016/s1001-0742(09)60122-4.
Li, L. et al., 2020: Air quality changes during the COVID-19 lockdown over the Yangtze River Delta Region: An insight into the impact of human activity pattern changes on air pollution variation. Science of the Total Environment, 732, 139282, doi: 10.1016/j.scitotenv.2020.139282.
Li, M. et al., 2018: Air quality co-benefits of carbon pricing in China. Nature Climate Change, 8(5), 398–403, doi: 10.1038/s41558-018-0139-4.
Li, M. et al., 2019: Persistent growth of anthropogenic non-methane volatile organic compound (NMVOC) emissions in China during 1990–2017: Drivers, speciation and ozone formation potential. Atmospheric Chemistry and Physics, 19(13), 8897–8913, doi: 10.5194/acp-19-8897-2019.
Li, Y. et al., 2016: Increasing importance of deposition of reduced nitrogen in the United States. Proceedings of the National Academy of Sciences, 113(21), 5874–5879, doi: 10.1073/pnas.1525736113.
Lin, J.T. et al., 2015: Influence of aerosols and surface reflectance on satellite NO2 retrieval: Seasonal and spatial characteristics and implications for NOx emission constraints. Atmospheric Chemistry and Physics, 15(19), 11217–11241, doi: 10.5194/acp-15-11217-2015.
Liu, F. et al., 2016: Recent reduction in NOx emissions over China: synthesis of satellite observations and emission inventories. Environmental Research Letters, 11(11), 114002, doi: 10.1088/1748-9326/11/11/114002.
Liu, H. et al., 2016: Health and climate impacts of ocean-going vessels in East Asia. Nature Climate Change, 6(11), 1037–1041, doi: 10.1038/nclimate3083.
Liu, L. et al., 2018: A PDRMIP Multimodel Study on the Impacts of Regional Aerosol Forcings on Global and Regional Precipitation. Journal of Climate, 31(11), 4429–4447, doi: 10.1175/jcli-d-17-0439.1.
Liu, L. et al., 2019: Estimating global surface ammonia concentrations inferred from satellite retrievals. Atmospheric Chemistry and Physics, 19(18), 12051–12066, doi: 10.5194/acp-19-12051-2019.
Liu, M. et al., 2018: Rapid SO2 emission reductions significantly increase tropospheric ammonia concentrations over the North China Plain. Atmospheric Chemistry and Physics, 18(24), 17933–17943, doi: 10.5194/acp-18-17933-2018.
Liu, Z. et al., 2020: Near-real-time monitoring of global CO2 emissions reveals the effects of the COVID-19 pandemic. Nature Communications, 11(1), 5172, doi: 10.1038/s41467-020-18922-7.
Llorens, L., J. Llusià, E.H. Murchie, J. Peñuelas, and D.J. Beerling, 2009: Monoterpene emissions and photoinhibition of “living fossil” trees grown under CO2 enrichment in a simulated Cretaceous polar environment. Journal of Geophysical Research: Biogeosciences, 114(G1), G01005, doi: 10.1029/2008jg000802.
Lombardozzi, D., J.P. Sparks, and G. Bonan, 2013: Integrating O3 influences on terrestrial processes: photosynthetic and stomatal response data available for regional and global modeling. Biogeosciences, 10(11), 6815–6831, doi: 10.5194/bg-10-6815-2013.
Lombardozzi, D., S. Levis, G. Bonan, P.G. Hess, and J.P. Sparks, 2015: The Influence of Chronic Ozone Exposure on Global Carbon and Water Cycles. Journal of Climate, 28(1), 292–305, doi: 10.1175/jcli-d-14-00223.1.
Lorente, A. et al., 2017: Structural uncertainty in air mass factor calculation for NO2 and HCHO satellite retrievals. Atmospheric Measurement Techniques, 10(3), 759–782, doi: 10.5194/amt-10-759-2017.
Loreto, F., M. Dicke, J.-P. Schnitzler, and T.C.J. Turlings, 2014: Plant volatiles and the environment. Plant, Cell & Environment, 37(8), 1905–1908, doi: 10.1111/pce.12369.
Loreto, F. et al., 2001: Monoterpene emission and monoterpene synthase activities in the Mediterranean evergreen oakQuercus ilexL. grown at elevated CO2 concentrations. Global Change Biology, 7(6), 709–717, doi: 10.1046/j.1354-1013.2001.00442.x.
Lowe, J.A. and D. Bernie, 2018: The impact of Earth system feedbacks on carbon budgets and climate response. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 376(2119), 20170263, doi: 10.1098/rsta.2017.0263.
Lu, X., M. Chen, Y. Liu, D.G. Miralles, and F. Wang, 2017: Enhanced water use efficiency in global terrestrial ecosystems under increasing aerosol loadings. Agricultural and Forest Meteorology, 237–238, 39–49, doi: 10.1016/j.agrformet.2017.02.002.
Lucas, P.L., D.P. van Vuuren, J.G.J. Olivier, and M.G.J. den Elzen, 2007: Long-term reduction potential of non-CO2 greenhouse gases. Environmental Science & Policy, 10(2), 85–103, doi: 10.1016/j.envsci.2006.10.007.
Lund, M.T., T.K. Berntsen, C. Heyes, Z. Klimont, and B.H. Samset, 2014: Global and regional climate impacts of black carbon and co-emitted species from the on-road diesel sector. Atmospheric Environment, 98, 50–58, doi: 10.1016/j.atmosenv.2014.08.033.
Lund, M.T. et al., 2012: Global-Mean Temperature Change from Shipping toward 2050: Improved Representation of the Indirect Aerosol Effect in Simple Climate Models. Environmental Science & Technology, 46(16), 8868–8877, doi: 10.1021/es301166e.
Lund, M.T. et al., 2017: Emission metrics for quantifying regional climate impacts of aviation. Earth System Dynamics, 8(3), 547–563, doi: 10.5194/esd-8-547-2017.
Lund, M.T. et al., 2018a: Concentrations and radiative forcing of anthropogenic aerosols from 1750 to 2014 simulated with the Oslo CTM3 and CEDS emission inventory. Geoscientific Model Development, 11(12), 4909–4931, doi: 10.5194/gmd-11-4909-2018.
Lund, M.T. et al., 2018b: Short Black Carbon lifetime inferred from a global set of aircraft observations. npj Climate and Atmospheric Science, 1(1), 31, doi: 10.1038/s41612-018-0040-x.
Lund, M.T. et al., 2020: A continued role of short-lived climate forcers under the Shared Socioeconomic Pathways. Earth System Dynamics, 11(4), 977–993, doi: 10.5194/esd-11-977-2020.
Luo, M. et al., 2015: Satellite observations of tropospheric ammonia and carbon monoxide: Global distributions, regional correlations and comparisons to model simulations. Atmospheric Environment, 106, 262–277, doi: 10.1016/j.atmosenv.2015.02.007.
MacMartin, D.G. et al., 2017: The Climate Response to Stratospheric Aerosol Geoengineering Can Be Tailored Using Multiple Injection Locations. Journal of Geophysical Research: Atmospheres, 122(23), 12574–12590, doi: 10.1002/2017jd026868.
Mahmood, R. et al., 2016: Seasonality of global and Arctic black carbon processes in the Arctic Monitoring and Assessment Programme models. Journal of Geophysical Research: Atmospheres, 121(12), 7100–7116, doi: 10.1002/2016jd024849.
Mahowald, N.M., 2011: Aerosol Indirect Effect on Biogeochemical Cycles and Climate. Science, 334(6057), 794–796, doi: 10.1126/science.1207374.
Mahowald, N.M. and C. Luo, 2003: A less dusty future?Geophysical Research Letters, 30(17), 1903, doi: 10.1029/2003gl017880.
Mahowald, N.M. et al., 2010: Observed 20th century desert dust variability: Impact on climate and biogeochemistry. Atmospheric Chemistry and Physics, 10(22), 10875–10893, doi: 10.5194/acp-10-10875-2010.
Mahowald, N.M. et al., 2017: Aerosol Deposition Impacts on Land and Ocean Carbon Cycles. Current Climate Change Reports, 3(1), 16–31, doi: 10.1007/s40641-017-0056-z.
Maione, M. et al., 2016: Air quality and climate change: Designing new win-win policies for Europe. Environmental Science & Policy, 65, 48–57, doi: 10.1016/j.envsci.2016.03.011.
Malavelle, F.F. et al., 2017: Strong constraints on aerosol–cloud interactions from volcanic eruptions. Nature, 546(7659), 485–491, doi: 10.1038/nature22974.
Malm, W.C., B.A. Schichtel, J.L. Hand, and J.L. Collett, 2017: Concurrent Temporal and Spatial Trends in Sulfate and Organic Mass Concentrations Measured in the IMPROVE Monitoring Program. Journal of Geophysical Research: Atmospheres, 122(19), 10462–10476, doi: 10.1002/2017jd026865.
Mangeon, S., R. Field, M. Fromm, C. McHugh, and A. Voulgarakis, 2015: Satellite versus ground-based estimates of burned area: A comparison between MODIS based burned area and fire agency reports over North America in 2007. The Anthropocene Review, 3(2), 76–92, doi: 10.1177/2053019615588790.
Mann, G.W. et al., 2014: Intercomparison and evaluation of global aerosol microphysical properties among AeroCom models of a range of complexity. Atmospheric Chemistry and Physics, 14(9), 4679–4713, doi: 10.5194/acp-14-4679-2014.
Marais, E.A. et al., 2018: Nitrogen oxides in the global upper troposphere: Interpreting cloud-sliced NO2 observations from the OMI satellite instrument. Atmospheric Chemistry and Physics, 18(23), 17017–17027, doi: 10.5194/acp-18-17017-2018.
Mariani, R.L. and W.Z. de Mello, 2007: PM2.5–10, PM2.5 and associated water-soluble inorganic species at a coastal urban site in the metropolitan region of Rio de Janeiro. Atmospheric Environment, 41(13), 2887–2892, doi: 10.1016/j.atmosenv.2006.12.009.
Markakis, K. et al., 2014: Air quality in the mid-21st century for the city of Paris under two climate scenarios; From the regional to local scale. Atmospheric Chemistry and Physics, 14(14), 7323–7340, doi: 10.5194/acp-14-7323-2014.
Markakis, K. et al., 2016: Mid-21st century air quality at the urban scale under the influence of changed climate and emissions – case studies for Paris and Stockholm. Atmospheric Chemistry and Physics, 16(4), 1877–1894, doi: 10.5194/acp-16-1877-2016.
Markandya, A. et al., 2018: Health co-benefits from air pollution and mitigation costs of the Paris Agreement: a modelling study. The Lancet Planetary Health, 2(3), e126–e133, doi: 10.1016/s2542-5196(18)30029-9.
Marlier, M.E., E.X. Bonilla, and L.J. Mickley, 2020: How Do Brazilian Fires Affect Air Pollution and Public Health?GeoHealth, 4(12), e2020GH000331, doi: 10.1029/2020gh000331.
Marlon, J.R. et al., 2016: Reconstructions of biomass burning from sediment-charcoal records to improve data–model comparisons. Biogeosciences, 13(11), 3225–3244, doi: 10.5194/bg-13-3225-2016.
Marsh, D.R., J.F. Lamarque, A.J. Conley, and L.M. Polvani, 2016: Stratospheric ozone chemistry feedbacks are not critical for the determination of climate sensitivity in CESM1(WACCM). Geophysical Research Letters, 43(8), 3928–3934, doi: 10.1002/2016gl068344.
Martin, S.T. et al., 2010: Sources and properties of Amazonian aerosol particles. Reviews of Geophysics, 48(2), RG2002, doi: 10.1029/2008rg000280.
Masiol, M. et al., 2015: Spatial, seasonal trends and transboundary transport of PM2.5 inorganic ions in the Veneto region (Northeastern Italy). Atmospheric Environment, 117, 19–31, doi: 10.1016/j.atmosenv. 2015.06.044.
McCollum, D.L. et al., 2013: Climate policies can help resolve energy security and air pollution challenges. Climatic Change, 119(2), 479–494, doi: 10.1007/s10584-013-0710-y.
McCollum, D.L. et al., 2018: Connecting the sustainable development goals by their energy inter-linkages. Environmental Research Letters, 13(3), 33006, doi: 10.1088/1748-9326/aaafe3.
McDonald, B.C. et al., 2018: Volatile chemical products emerging as largest petrochemical source of urban organic emissions. Science, 359(6377), 760–764, doi: 10.1126/science.aaq0524.
McDuffie, E.E. et al., 2020: A global anthropogenic emission inventory of atmospheric pollutants from sector- and fuel-specific sources (1970–2017): an application of the Community Emissions Data System (CEDS). Earth System Science Data, 12(4), 3413–3442, doi: 10.5194/essd-12-3413-2020.
McFiggans, G. et al., 2019: Secondary organic aerosol reduced by mixture of atmospheric vapours. Nature, 565(7741), 587–593, doi: 10.1038/s41586-018-0871-y.
McNorton, J. et al., 2016: Role of OH variability in the stalling of the global atmospheric CH4 growth rate from 1999 to 2006. Atmospheric Chemistry and Physics, 16(12), 7943–7956, doi: 10.5194/acp-16-7943-2016.
McNorton, J. et al., 2018: Attribution of recent increases in atmospheric methane through 3-D inverse modelling. Atmospheric Chemistry and Physics, 18(24), 18149–18168, doi: 10.5194/acp-18-18149-2018.
Medici, G., K.L. Cummins, D.J. Cecil, W.J. Koshak, and S.D. Rudlosky, 2017: The Intracloud Lightning Fraction in the Contiguous United States. Monthly Weather Review, 145(11), 4481–4499, doi: 10.1175/mwr-d-16-0426.1.
Meehl, G.A. et al., 2020: Context for interpreting equilibrium climate sensitivity and transient climate response from the CMIP6 Earth system models. Science Advances, 6(26), eaba1981, doi: 10.1126/sciadv.aba1981.
Megaritis, A.G., C. Fountoukis, P.E. Charalampidis, C. Pilinis, and S.N. Pandis, 2013: Response of fine particulate matter concentrations to changes of emissions and temperature in Europe. Atmospheric Chemistry and Physics, 13(6), 3423–3443, doi: 10.5194/acp-13-3423-2013.
Meinshausen, M. et al., 2011: The RCP greenhouse gas concentrations and their extensions from 1765 to 2300. Climatic Change, 109(1–2), 213–241, doi: 10.1007/s10584-011-0156-z.
Meinshausen, M. et al., 2020: The shared socio-economic pathway (SSP) greenhouse gas concentrations and their extensions to 2500. Geoscientific Model Development, 13(8), 3571–3605, doi: 10.5194/gmd-13-3571-2020.
Melamed, M.L., J. Schmale, and E. von Schneidemesser, 2016: Sustainable policy – key considerations for air quality and climate change. Current Opinion in Environmental Sustainability, 23, 85–91, doi: 10.1016/j.cosust.2016.12.003.
Mercado, L.M. et al., 2009: Impact of changes in diffuse radiation on the global land carbon sink. Nature, 458(7241), 1014–1017, doi: 10.1038/nature07949.
Meredith, M. et al., 2019: Polar Regions. In: IPCC Special Report on the Ocean and Cryosphere in a Changing Climate[Pörtner, H.-O., D.C. Roberts, V. Masson-Delmotte, P. Zhai, M. Tignor, E. Poloczanska, K. Mintenbeck, A. Alegría, M. Nicolai, A. Okem, J. Petzold, B. Rama, and N.M. Weyer (eds.)]. In Press, pp. 203–320, www.ipcc.ch/srocc/chapter/chapter-3-2.
Mertens, M., A. Kerkweg, V. Grewe, P. Jöckel, and R. Sausen, 2020: Attributing ozone and its precursors to land transport emissions in Europe and Germany. Atmospheric Chemistry and Physics, 20(13), 7843–7873, doi: 10.5194/acp-20-7843-2020.
Messina, P. et al., 2016: Global biogenic volatile organic compound emissions in the ORCHIDEE and MEGAN models and sensitivity to key parameters. Atmospheric Chemistry and Physics, 16(22), 14169–14202, doi: 10.5194/acp-16-14169-2016.
Meul, S., U. Langematz, P. Kröger, S. Oberländer-Hayn, and P. Jöckel, 2018: Future changes in the stratosphere-to-troposphere ozone mass flux and the contribution from climate change and ozone recovery. Atmospheric Chemistry and Physics, 18(10), 7721–7738, doi: 10.5194/acp-18-7721-2018.
Miller, D.J., K. Sun, L. Tao, M.A. Khan, and M.A. Zondlo, 2014: Open-path, quantum cascade-laser-based sensor for high-resolution atmospheric ammonia measurements. Atmospheric Measurement Techniques, 7(1), 81–93, doi: 10.5194/amt-7-81-2014.
Miller, S.M. et al., 2019: China’s coal mine methane regulations have not curbed growing emissions. Nature Communications, 10(1), 303, doi: 10.1038/s41467-018-07891-7.
Mills, G. et al., 2016: Ozone impacts on vegetation in a nitrogen enriched and changing climate. Environmental Pollution, 208, 898–908, doi: 10.1016/j.envpol.2015.09.038.
Ming, Y. et al., 2021: Assessing the Influence of COVID-19 on the Shortwave Radiative Fluxes Over the East Asian Marginal Seas. Geophysical Research Letters, 48(3), e2020GL091699, doi: 10.1029/2020gl091699.
Mitchell, D.L. and W. Finnegan, 2009: Modification of cirrus clouds to reduce global warming. Environmental Research Letters, 4(4), 045102, doi: 10.1088/1748-9326/4/4/045102.
Miyazaki, K., H.J. Eskes, and K. Sudo, 2015: A tropospheric chemistry reanalysis for the years 2005–2012 based on an assimilation of OMI, MLS, TES, and MOPITT satellite data. Atmospheric Chemistry and Physics, 15(14), 8315–8348, doi: 10.5194/acp-15-8315-2015.
Miyazaki, K., H.J. Eskes, K. Sudo, and C. Zhang, 2014: Global lightning NOx production estimated by an assimilation of multiple satellite data sets. Atmospheric Chemistry and Physics, 14(7), 3277–3305, doi: 10.5194/acp-14-3277-2014.
Miyazaki, K. et al., 2017: Decadal changes in global surface NOx emissions from multi-constituent satellite data assimilation. Atmospheric Chemistry and Physics, 17(2), 807–837, doi: 10.5194/acp-17-807-2017.
Mkoma, S., 2008: Physico-Chemical Characterisation of Atmospheric Aerosols in Tanzania, with Emphasis on the Carbonaceous Aerosol Components and on Chemical Mass Closure., Ghent University, Faculty of Sciences, Ghent, Belgium, 182 pp., doi: 1854/9881.
Mkoma, S.L., W. Maenhaut, X. Chi, W. Wang, and N. Raes, 2009: Characterisation of PM10 atmospheric aerosols for the wet season 2005 at two sites in East Africa. Atmospheric Environment, 43(3), 631–639, doi: 10.1016/j.atmosenv.2008.10.008.
Mohankumar Sajeev, E.P., W. Winiwarter, and B. Amon, 2018: Greenhouse Gas and Ammonia Emissions from Different Stages of Liquid Manure Management Chains: Abatement Options and Emission Interactions. Journal of Environmental Quality, 47(1), 30–41, doi: 10.2134/jeq2017.05.0199.
Molina, L.T. et al., 2007: Air quality in North America’s most populous city – overview of the MCMA-2003 campaign. Atmospheric Chemistry and Physics, 7(10), 2447–2473, doi: 10.5194/acp-7-2447-2007.
Molina, L.T. et al., 2010: An overview of the MILAGRO 2006 Campaign: Mexico City emissions and their transport and transformation. Atmospheric Chemistry and Physics, 10(18), 8697–8760, doi: 10.5194/acp-10-8697-2010.
Monks, S.A. et al., 2015: Multi-model study of chemical and physical controls on transport of anthropogenic and biomass burning pollution to the Arctic. Atmospheric Chemistry and Physics, 15(6), 3575–3603, doi: 10.5194/acp-15-3575-2015.
Monteiro dos Santos, D.A., J.F. Brito, J.M. Godoy, and P. Artaxo, 2016: Ambient concentrations and insights on organic and elemental carbon dynamics in São Paulo, Brazil. Atmospheric Environment, 144, 226–233, doi: 10.1016/j.atmosenv.2016.08.081.
Montzka, S.A. et al., 2011: Small Interannual Variability of Global Atmospheric Hydroxyl. Science, 331(6013), 67–69, doi: 10.1126/science.1197640.
Montzka, S.A. et al., 2015: Recent Trends in Global Emissions of Hydrochlorofluorocarbons and Hydrofluorocarbons: Reflecting on the 2007 Adjustments to the Montreal Protocol. The Journal of Physical Chemistry A, 119(19), 4439–4449, doi: 10.1021/jp5097376.
Morgan, W.T. et al., 2020: Transformation and ageing of biomass burning carbonaceous aerosol over tropical South America from aircraft in situ measurements during SAMBBA. Atmospheric Chemistry and Physics, 20(9), 5309–5326, doi: 10.5194/acp-20-5309-2020.
Morgenstern, O. et al., 2017: Review of the global models used within phase 1 of the Chemistry–Climate Model Initiative (CCMI). Geoscientific Model Development, 10(2), 639–671, doi: 10.5194/gmd-10-639-2017.
Morgenstern, O. et al., 2018: Ozone sensitivity to varying greenhouse gases and ozone-depleting substances in CCMI-1 simulations. Atmospheric Chemistry and Physics, 18(2), 1091–1114, doi: 10.5194/acp-18-1091-2018.
Morgenstern, O. et al., 2020: Reappraisal of the Climate Impacts of Ozone-Depleting Substances. Geophysical Research Letters, 47(20), e2020GL088295, doi: 10.1029/2020gl088295.
Mortier, A. et al., 2020: Evaluation of climate model aerosol trends with ground-based observations over the last 2 decades – an AeroCom and CMIP6 analysis. Atmospheric Chemistry and Physics, 20(21), 13355–13378, doi: 10.5194/acp-20-13355-2020.
Moteki, N., T. Mori, H. Matsui, and S. Ohata, 2019: Observational constraint of in-cloud supersaturation for simulations of aerosol rainout in atmospheric models. npj Climate and Atmospheric Science, 2(1), 6, doi: 10.1038/s41612-019-0063-y.
Motos, G. et al., 2019: Cloud droplet activation properties and scavenged fraction of black carbon in liquid-phase clouds at the high-alpine research station Jungfraujoch (3580 m a.s.l.). Atmospheric Chemistry and Physics, 19(6), 3833–3855, doi: 10.5194/acp-19-3833-2019.
Mülmenstädt, J. and G. Feingold, 2018: The Radiative Forcing of Aerosol–Cloud Interactions in Liquid Clouds: Wrestling and Embracing Uncertainty. Current Climate Change Reports, 4(1), 23–40, doi: 10.1007/s40641-018-0089-y.
Muri, H. et al., 2018: Climate Response to Aerosol Geoengineering: A Multimethod Comparison. Journal of Climate, 31(16), 6319–6340, doi: 10.1175/jcli-d-17-0620.1.
Murray, L.T., 2016: Lightning NOx and Impacts on Air Quality. Current Pollution Reports, 2(2), 115–133, doi: 10.1007/s40726-016-0031-7.
Murray, L.T., J.A. Logan, and D.J. Jacob, 2013: Interannual variability in tropical tropospheric ozone and OH: The role of lightning. Journal of Geophysical Research: Atmospheres, 118(19), 11468–11480, doi: 10.1002/jgrd.50857.
Murray, L.T. et al., 2014: Factors controlling variability in the oxidative capacity of the troposphere since the Last Glacial Maximum. Atmospheric Chemistry and Physics, 14(7), 3589–3622, doi: 10.5194/acp-14-3589-2014.
Muthers, S. et al., 2014: The coupled atmosphere–chemistry–ocean model SOCOL-MPIOM. Geoscientific Model Development, 7(5), 2157–2179, doi: 10.5194/gmd-7-2157-2014.
Myhre, G., T.F. Berglen, C.E.L. Myhre, and I.S.A. Isaksen, 2004: The radiative effect of the anthropogenic influence on the stratospheric sulfate aerosol layer. Tellus B: Chemical and Physical Meteorology, 56(3), 294–299, doi: 10.3402/tellusb.v56i3.16431.
Myhre, G. et al., 2013: Anthropogenic and Natural Radiative Forcing. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change[Stocker, T.F., D. Qin, G.-K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P.M. Midgley (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 659–740, doi: 10.1017/cbo9781107415324.018.
Myhre, G. et al., 2017: Multi-model simulations of aerosol and ozone radiative forcing due to anthropogenic emission changes during the period 1990–2015. Atmospheric Chemistry and Physics, 17(4), 2709–2720, doi: 10.5194/acp-17-2709-2017.
Myriokefalitakis, S. et al., 2016: Ozone and carbon monoxide budgets over the Eastern Mediterranean. Science of the Total Environment, 563–564, 40–52, doi: 10.1016/j.scitotenv.2016.04.061.
Naik, V. et al., 2013: Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmospheric Chemistry and Physics, 13(10), 5277–5298, doi: 10.5194/acp-13-5277-2013.
Nault, B.A. et al., 2017: Lightning NOx emissions: Reconciling Measured and Modeled Estimates With Updated NOx Chemistry. Geophysical Research Letters, 44(18), 9479–9488, doi: 10.1002/2017gl074436.
Naus, S. et al., 2019: Constraints and biases in a tropospheric two-box model of OH. Atmospheric Chemistry and Physics, 19(1), 407–424, doi: 10.5194/acp-19-407-2019.
Nazarenko, L. et al., 2017: Interactive nature of climate change and aerosol forcing. Journal of Geophysical Research: Atmospheres, 122(6), 3457–3480, doi: 10.1002/2016jd025809.
Nenes, A., S.N. Pandis, R.J. Weber, and A. Russell, 2020: Aerosol pH and liquid water content determine when particulate matter is sensitive to ammonia and nitrate availability. Atmospheric Chemistry and Physics, 20(5), 3249–3258, doi: 10.5194/acp-20-3249-2020.
Nguyen, G.T.H., H. Shimadera, K. Uranishi, T. Matsuo, and A. Kondo, 2019: Numerical assessment of PM2.5 and O3 air quality in Continental Southeast Asia: Impacts of potential future climate change. Atmospheric Environment, 215, 116901, doi: 10.1016/j.atmosenv.2019.116901.
Nguyen, T.L., L. Vereecken, and J. Peeters, 2010: HOxRegeneration in the Oxidation of Isoprene III: Theoretical Study of the key Isomerisation of the Z- δ-hydroxy-peroxy Isoprene Radicals. ChemPhysChem, 11(18), 3996–4001, doi: 10.1002/cphc.201000480.
Nicely, J.M. et al., 2017: Quantifying the causes of differences in tropospheric OH within global models. Journal of Geophysical Research: Atmospheres, 122(3), 1983–2007, doi: 10.1002/2016jd026239.
Nicely, J.M. et al., 2018: Changes in Global Tropospheric OH Expected as a Result of Climate Change Over the Last Several Decades. Journal of Geophysical Research: Atmospheres, 123(18), 10774–10795, doi: 10. 1029/2018jd028388.
Nicewonger, M.R., K.R. Verhulst, M. Aydin, and E.S. Saltzman, 2016: Preindustrial atmospheric ethane levels inferred from polar ice cores: A constraint on the geologic sources of atmospheric ethane and methane. Geophysical Research Letters, 43(1), 214–221, doi: 10.1002/ 2015gl066854.
Nicewonger, M.R., M. Aydin, M.J. Prather, and E.S. Saltzman, 2018: Large changes in biomass burning over the last millennium inferred from paleoatmospheric ethane in polar ice cores. Proceedings of the National Academy of Sciences, 115(49), 12413–12418, doi: 10.1073/pnas.1807172115.
Niemeier, U. and C. Timmreck, 2015: What is the limit of climate engineering by stratospheric injection of SO2?Atmospheric Chemistry and Physics, 15(16), 9129–9141, doi: 10.5194/acp-15-9129-2015.
Niemeier, U. and H. Schmidt, 2017: Changing transport processes in the stratosphere by radiative heating of sulfate aerosols. Atmospheric Chemistry and Physics, 17(24), 14871–14886, doi: 10.5194/acp-17-14871-2017.
Niinemets, Ü, S. Fares, P. Hardley, and K.J. Jardine, 2014: Bidirectional exchange of biogenic volatiles with vegetation: emission sources, reactions, breakdown and deposition. Plant, Cell & Environment, 37(8), 1790–1809, doi: 10.1111/pce.12322.
Nolte, C.G., T.L. Spero, J.H. Bowden, M.S. Mallard, and P.D. Dolwick, 2018: The potential effects of climate change on air quality across the conterminous US at 2030 under three Representative Concentration Pathways. Atmospheric Chemistry and Physics, 18(20), 15471–15489, doi: 10.5194/acp-18-15471-2018.
Nowack, P.J. et al., 2015: A large ozone-circulation feedback and its implications for global warming assessments. Nature Climate Change, 5(1), 41–45, doi: 10.1038/nclimate2451.
NRC, 2015: Climate Intervention: Reflecting Sunlight to Cool Earth. National Research Council (NRC). The National Academies Press, Washington, DC, USA, 260 pp., doi: 10.17226/18988.
O’Connor, F.M. et al., 2021: Assessment of pre-industrial to present-day anthropogenic effective radiative forcing in UKESM1. Atmospheric Chemistry and Physics, 21, 1211–1243, doi: 10.5194/acp-21-1211-2021.
O’Neill, B.C. et al., 2016: The Scenario Model Intercomparison Project (ScenarioMIP) for CMIP6. Geoscientific Model Development, 9(9), 3461–3482, doi: 10.5194/gmd-9-3461-2016.
Oberländer-Hayn, S., S. Meul, U. Langematz, J. Abalichin, and F. Haenel, 2015: A chemistry–climate model study of past changes in the Brewer–Dobson circulation. Journal of Geophysical Research: Atmospheres, 120(14), 6742–6757, doi: 10.1002/2014jd022843.
Ohata, S., N. Moteki, T. Mori, M. Koike, and Y. Kondo, 2016: A key process controlling the wet removal of aerosols: New observational evidence. Scientific Reports, 6(1), 1–9, doi: 10.1038/srep34113.
Oliveira, P.H.F. et al., 2007: The effects of biomass burning aerosols and clouds on the CO2 flux in Amazonia. Tellus B: Chemical and Physical Meteorology, 59(3), 338–349, doi: 10.1111/j.1600-0889.2007.00270.x.
Oliver, R.J. et al., 2018: Large but decreasing effect of ozone on the European carbon sink. Biogeosciences, 15(13), 4245–4269, doi: 10.5194/bg-15-4245-2018.
Olivié, D.J.L. and G.P. Peters, 2013: Variation in emission metrics due to variation in CO2 and temperature impulse response functions. Earth System Dynamics, 4(2), 267–286, doi: 10.5194/esd-4-267-2013.
Oram, D.E. et al., 2017: A growing threat to the ozone layer from short-lived anthropogenic chlorocarbons. Atmospheric Chemistry and Physics, 17(19), 11929–11941, doi: 10.5194/acp-17-11929-2017.
Orbe, C. et al., 2018: Large-scale tropospheric transport in the Chemistry–Climate Model Initiative (CCMI) simulations. Atmospheric Chemistry and Physics, 18(10), 7217–7235, doi: 10.5194/acp-18-7217-2018.
Oswald, E.M., L.A. Dupigny-Giroux, E.M. Leibensperger, R. Poirot, and J. Merrell, 2015: Climate controls on air quality in the Northeastern U.S.: An examination of summertime ozone statistics during 1993–2012. Atmospheric Environment, 112, 278–288, doi: 10.1016/j.atmosenv.2015.04.019.
Ovadnevaite, J. et al., 2014: A sea spray aerosol flux parameterization encapsulating wave state. Atmospheric Chemistry and Physics, 14(4), 1837–1852, doi: 10.5194/acp-14-1837-2014.
Paasonen, P. et al., 2013: Warming-induced increase in aerosol number concentration likely to moderate climate change. Nature Geoscience, 6(6), 438–442, doi: 10.1038/ngeo1800.
Pacifico, F., G.A. Folberth, C.D. Jones, S.P. Harrison, and W.J. Collins, 2012: Sensitivity of biogenic isoprene emissions to past, present, and future environmental conditions and implications for atmospheric chemistry. Journal of Geophysical Research: Atmospheres, 117(D22), D22302, doi: 10.1029/2012jd018276.
Palmer, P.I. et al., 2006: Quantifying the seasonal and interannual variability of North American isoprene emissions using satellite observations of the formaldehyde column. Journal of Geophysical Research: Atmospheres, 111(D12), D12315, doi: 10.1029/2005jd006689.
Pan, Y. et al., 2018: Identifying Ammonia Hotspots in China Using a National Observation Network. Environmental Science & Technology, 52(7), 3926–3934, doi: 10.1021/acs.est.7b05235.
Pant, P. et al., 2015: Characterization of ambient PM2.5 at a pollution hotspot in New Delhi, India and inference of sources. Atmospheric Environment, 109, 178–189, doi: 10.1016/j.atmosenv.2015.02.074.
Papanastasiou, D.K., A. Beltrone, P. Marshall, and J.B. Burkholder, 2018: Global warming potential estimates for the C1-C3hydrochlorofluorocarbons (HCFCs) included in the Kigali Amendment to the Montreal Protocol. Atmospheric Chemistry and Physics, 18(9), 6317–6330, doi: 10.5194/ acp-18-6317-2018.
Parrish, D.D., H.B. Singh, L. Molina, and S. Madronich, 2011: Air quality progress in North American megacities: A review. Atmospheric Environment, 45(39), 7015–7025, doi: 10.1016/j.atmosenv.2011.09.039.
Partanen, A.-I. et al., 2012: Direct and indirect effects of sea spray geoengineering and the role of injected particle size. Journal of Geophysical Research: Atmospheres, 117(D2), D02203, doi: 10.1029/2011jd016428.
Partanen, A.-I. et al., 2013: Climate and air quality trade-offs in altering ship fuel sulfur content. Atmospheric Chemistry and Physics, 13(23), 12059–12071, doi: 10.5194/acp-13-12059-2013.
Pathak, R.K., T. Wang, K.F. Ho, and S.C. Lee, 2011: Characteristics of summertime PM2.5 organic and elemental carbon in four major Chinese cities: Implications of high acidity for water-soluble organic carbon (WSOC). Atmospheric Environment, 45(2), 318–325, doi: 10.1016/j.atmosenv.2010.10.021.
Patra, P.K. et al., 2021: Methyl Chloroform Continues to Constrain the Hydroxyl (OH) Variability in the Troposphere. Journal of Geophysical Research: Atmospheres, 126(4), e2020JD033862, doi: 10.1029/2020jd033862.
Patris, N. et al., 2002: First sulfur isotope measurements in central Greenland ice cores along the preindustrial and industrial periods. Journal of Geophysical Research: Atmospheres, 107(D11), ACH 6–1–ACH 6–11, doi: 10.1029/2001jd000672.
Paulot, F., D. Paynter, P. Ginoux, V. Naik, and L.W. Horowitz, 2018a: Changes in the aerosol direct radiative forcing from 2001 to 2015: Observational constraints and regional mechanisms. Atmospheric Chemistry and Physics, 18(17), 13265–13281, doi: 10.5194/acp-18-13265-2018.
Paulot, F. et al., 2009: Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science, 325(5941), 730–733, doi: 10.1126/science.1172910.
Paulot, F. et al., 2014: Ammonia emissions in the United States, European Union, and China derived by high-resolution inversion of ammonium wet deposition data: Interpretation with a new agricultural emissions inventory (MASAGE_NH3). Journal of Geophysical Research: Atmospheres, 119(7), 4343–4364, doi: 10.1002/2013jd021130.
Paulot, F. et al., 2016: Sensitivity of nitrate aerosols to ammonia emissions and to nitrate chemistry: Implications for present and future nitrate optical depth. Atmospheric Chemistry and Physics, 16(3), 1459–1477, doi: 10.5194/acp-16-1459-2016.
Paulot, F. et al., 2018b: Representing sub-grid scale variations in nitrogen deposition associated with land use in a global Earth system model: Implications for present and future nitrogen deposition fluxes over North America. Atmospheric Chemistry and Physics, 18(24), 17963–17978, doi: 10.5194/acp-18-17963-2018.
Paulot, F. et al., 2020: Revisiting the Impact of Sea Salt on Climate Sensitivity. Geophysical Research Letters, 47(3), e2019GL085601, doi: 10.1029/2019gl085601.
Peeters, J., T.L. Nguyen, and L. Vereecken, 2009: HOxradical regeneration in the oxidation of isoprene. Physical Chemistry Chemical Physics, 11(28), 5935–5939, doi: 10.1039/b908511d.
Peeters, J., J.F. Müller, T. Stavrakou, and V.S. Nguyen, 2014: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism. Journal of Physical Chemistry A, 118(38), 8625–8643, doi: 10.1021/jp5033146.
Peng, S. et al., 2016: Inventory of anthropogenic methane emissions in mainland China from 1980 to 2010. Atmospheric Chemistry and Physics, 16(22), 14545–14562, doi: 10.5194/acp-16-14545-2016.
Peng, W., J. Yang, F. Wagner, and D.L. Mauzerall, 2017: Substantial air quality and climate co-benefits achievable now with sectoral mitigation strategies in China. Science of the Total Environment, 598, 1076–1084, doi: 10.1016/j.scitotenv.2017.03.287.
Penner, J.E., C. Zhou, and X. Liu, 2015: Can cirrus cloud seeding be used for geoengineering?Geophysical Research Letters, 42(20), 8775–8782, doi: 10.1002/2015gl065992.
Penrod, A., Y. Zhang, K. Wang, S.Y. Wu, and L.R. Leung, 2014: Impacts of future climate and emission changes on U.S. air quality. Atmospheric Environment, 89, 533–547, doi: 10.1016/j.atmosenv.2014.01.001.
Peñuelas, J. and M. Staudt, 2010: BVOCs and global change. Trends in Plant Science, 15(3), 133–144, doi: 10.1016/j.tplants.2009.12.005.
Persad, G.G. and K. Caldeira, 2018: Divergent global-scale temperature effects from identical aerosols emitted in different regions. Nature Communications, 9(1), 3289, doi: 10.1038/s41467-018-05838-6.
Petetin, H. et al., 2018: Representativeness of the IAGOS airborne measurements in the lower troposphere. Elementa: Science of the Anthropocene, 6, 23, doi: 10.1525/elementa.280.
Petit, J.-E. et al., 2015: Two years of near real-time chemical composition of submicron aerosols in the region of Paris using an Aerosol Chemical Speciation Monitor (ACSM) and a multi-wavelength Aethalometer. Atmospheric Chemistry and Physics, 15(6), 2985–3005, doi: 10.5194/acp-15-2985-2015.
Petrenko, V. et al., 2013: A 60 yr record of atmospheric carbon monoxide reconstructed from Greenland firn air. Atmospheric Chemistry and Physics, 13(15), 7567–7585, doi: 10.5194/acp-13-7567-2013.
Pétron, G., A.M. Crotwell, E. Dlugokencky, and J.W. Mund, 2019: Atmospheric carbon monoxide dry air mole fractions from the NOAA ESRL carbon cycle cooperative global air sampling network, 1988–2017, Version: 2019–08. National Oceanic & Atmospheric Administration (NOAA) Earth System Research Laboratory (ESRL), Boulder, CO, USA. Retrieved from: ftp://aftp.cmdl.noaa.gov/data/trace_gases/co/flask/surface.
Pétron, G. et al., 2014: A new look at methane and nonmethane hydrocarbon emissions from oil and natural gas operations in the Colorado Denver-Julesburg Basin. Journal of Geophysical Research: Atmospheres, 119(11), 6836–6852, doi: 10.1002/2013jd021272.
Petzold, A. et al., 2013: Recommendations for reporting black carbon measurements. Atmospheric Chemistry and Physics, 13(16), 8365–8379, doi: 10.5194/acp-13-8365-2013.
Pierce, J.R., D.K. Weisenstein, P. Heckendorn, T. Peter, and D.W. Keith, 2010: Efficient formation of stratospheric aerosol for climate engineering by emission of condensible vapor from aircraft. Geophysical Research Letters, 37(18), L18805, doi: 10.1029/2010gl043975.
Pierrehumbert, R.T., 2014: Short-Lived Climate Pollution. Annual Review of Earth and Planetary Sciences, 42(1), 341–379, doi: 10.1146/annurev-earth-060313-054843.
Pinder, R.W., P.J. Adams, and S.N. Pandis, 2007: Ammonia Emission Controls as a Cost-Effective Strategy for Reducing Atmospheric Particulate Matter in the Eastern United States. Environmental Science & Technology, 41(2), 380–386, doi: 10.1021/es060379a.
Pommier, M. et al., 2018: Impact of regional climate change and future emission scenarios on surface O3 and PM2.5 over India. Atmospheric Chemistry and Physics, 18(1), 103–127, doi: 10.5194/acp-18-103-2018.
Ponater, M., S. Marquart, R. Sausen, and U. Schumann, 2005: On contrail climate sensitivity. Geophysical Research Letters, 32(10), L10706, doi: 10.1029/2005gl022580.
Porter, W.C., C.L. Heald, D. Cooley, and B. Russell, 2015: Investigating the observed sensitivities of air-quality extremes to meteorological drivers via quantile regression. Atmospheric Chemistry and Physics, 15(18), 10349–10366, doi: 10.5194/acp-15-10349-2015.
Possell, M., C. Nicholas Hewitt, and D.J. Beerling, 2005: The effects of glacial atmospheric CO2 concentrations and climate on isoprene emissions by vascular plants. Global Change Biology, 11(1), 60–69, doi: 10.1111/j.1365-2486.2004.00889.x.
Potosnak, M.J. et al., 2014: Observed and modeled ecosystem isoprene fluxes from an oak-dominated temperate forest and the influence of drought stress. Atmospheric Environment, 84, 314–322, doi: 10.1016/j.atmosenv.2013.11.055.
Prather, M.J., 1994: Lifetimes and eigenstates in atmospheric chemistry. Geophysical Research Letters, 21(9), 801–804, doi: 10.1029/94gl00840.
Prather, M.J., 1996: Time scales in atmospheric chemistry: Theory, GWPs for CH4 and CO, and runaway growth. Geophysical Research Letters, 23(19), 2597–2600, doi: 10.1029/96gl02371.
Prather, M.J. and C.D. Holmes, 2017: Overexplaining or underexplaining methane’s role in climate change. Proceedings of the National Academy of Sciences, 114(21), 5324–5326, doi: 10.1073/pnas.1704884114.
Prather, M.J., C.D. Holmes, and J. Hsu, 2012: Reactive greenhouse gas scenarios: Systematic exploration of uncertainties and the role of atmospheric chemistry. Geophysical Research Letters, 39(9), L09803, doi: 10.1029/2012gl051440.
Prather, M.J. et al., 2001: Atmospheric Chemistry and Greenhouse Gases. In: Climate Change 2001: The Physical Science Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change[Y. Ding, D.J. Griggs, M. Noguer, P.J. van der Linden, X. Dai, K. Maskell, and C.A. Johnson (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 239–287, www.ipcc.ch/report/ar3/wg1.
Price, C. and D. Rind, 1992: A simple lightning parameterization for calculating global lightning distributions. Journal of Geophysical Research: Atmospheres, 97(D9), 9919–9933, doi: 10.1029/92jd00719.
Pringle, K.J. et al., 2012: A multi-model assessment of the impact of sea spray geoengineering on cloud droplet number. Atmospheric Chemistry and Physics, 12(23), 11647–11663, doi: 10.5194/acp-12-11647-2012.
Prinn, R.G. et al., 2018: History of chemically and radiatively important atmospheric gases from the Advanced Global Atmospheric Gases Experiment (AGAGE). Earth System Science Data, 10(2), 985–1018, doi: 10.5194/essd-10-985-2018.
Purohit, P. and L. Höglund-Isaksson, 2017: Global emissions of fluorinated greenhouse gases 2005-2050 with abatement potentials and costs. Atmospheric Chemistry and Physics, 17(4), 2795–2816, doi: 10.5194/acp-17-2795-2017.
Purohit, P. et al., 2020: Electricity savings and greenhouse gas emission reductions from global phase-down of hydrofluorocarbons. Atmospheric Chemistry and Physics, 20(19), 11305–11327, doi: 10.5194/acp- 20-11305-2020.
Pusede, S.E. et al., 2016: On the effectiveness of nitrogen oxide reductions as a control over ammonium nitrate aerosol. Atmospheric Chemistry and Physics, 16(4), 2575–2596, doi: 10.5194/acp-16-2575-2016.
Putaud, J.-P. et al., 2010: A European aerosol phenomenology – 3: Physical and chemical characteristics of particulate matter from 60 rural, urban, and kerbside sites across Europe. Atmospheric Environment, 44(10), 1308–1320, doi: 10.1016/j.atmosenv.2009.12.011.
Qiao, X. et al., 2018: Source apportionment of PM2.5 for 25 Chinese provincial capitals and municipalities using a source-oriented Community Multiscale Air Quality model. Science of The Total Environment, 612, 462–471, doi: 10.1016/j.scitotenv.2017.08.272.
Querol, X. et al., 2013: Variability of carbonaceous aerosols in remote, rural, urban and industrial environments in Spain: Implications for air quality policy. Atmospheric Chemistry and Physics, 13(13), 6185–6206, doi: 10.5194/acp-13-6185-2013.
Quiquet, A. et al., 2015: The relative importance of methane sources and sinks over the Last Interglacial period and into the last glaciation. Quaternary Science Reviews, 112, 1–16, doi: 10.1016/j.quascirev.2015.01.004.
Radhi, M. et al., 2010: Optical, physical and chemical characteristics of Australian continental aerosols: results from a field experiment. Atmospheric Chemistry and Physics, 10(13), 5925–5942, doi: 10.5194/acp-10-5925-2010.
Rafaj, P. and M. Amann, 2018: Decomposing air pollutant emissions in Asia: Determinants and projections. Energies, 11(5), 1299, doi: 10.3390/en11051299.
Rafaj, P., M. Amann, J. Siri, and H. Wuester, 2014: Changes in European greenhouse gas and air pollutant emissions 1960–2010: decomposition of determining factors. Climatic Change, 124(3), 477–504, doi: 10.1007/s10584-013-0826-0.
Rafaj, P. et al., 2018: Outlook for clean air in the context of sustainable development goals. Global Environmental Change, 53, 1–11, doi: 10.1016/j.gloenvcha.2018.08.008.
Randerson, J.T., Y. Chen, G.R. van der Werf, B.M. Rogers, and D.C. Morton, 2012: Global burned area and biomass burning emissions from small fires. Journal of Geophysical Research: Biogeosciences, 117(G4), G04012, doi: 10.1029/2012jg002128.
Randles, C.A. et al., 2017: The MERRA-2 aerosol reanalysis, 1980 onward. Part I: System description and data assimilation evaluation. Journal of Climate, 30(17), 6823–6850, doi: 10.1175/jcli-d-16-0609.1.
Rao, S. et al., 2013: Better air for better health: Forging synergies in policies for energy access, climate change and air pollution. Global Environmental Change, 23(5), 1122–1130, doi: 10.1016/j.gloenvcha.2013.05.003.
Rao, S. et al., 2016: A multi-model assessment of the co-benefits of climate mitigation for global air quality. Environmental Research Letters, 11(12), 124013, doi: 10.1088/1748-9326/11/12/124013.
Rao, S. et al., 2017: Future air pollution in the Shared Socio-economic Pathways. Global Environmental Change, 42, 346–358, doi: 10.1016/j.gloenvcha.2016.05.012.
Rap, A., P.M. Forster, J.M. Haywood, A. Jones, and O. Boucher, 2010: Estimating the climate impact of linear contrails using the UK Met Office climate model. Geophysical Research Letters, 37(20), L20703, doi: 10.1029/2010gl045161.
Rap, A. et al., 2015: Fires increase Amazon forest productivity through increases in diffuse radiation. Geophysical Research Letters, 42(11), 4654–4662, doi: 10.1002/2015gl063719.
Rasch, P.J., P.J. Crutzen, and D.B. Coleman, 2008: Exploring the geoengineering of climate using stratospheric sulfate aerosols: The role of particle size. Geophysical Research Letters, 35(2), L02809, doi: 10.1029/ 2007gl032179.
Rasch, P.J., J. Latham, and C.-C.J. Chen, 2009: Geoengineering by cloud seeding: influence on sea ice and climate system. Environmental Research Letters, 4(4), 045112, doi: 10.1088/1748-9326/4/4/045112.
Reddington, C.L. et al., 2017: The global aerosol synthesis and science project (GASSP): Measurements and modeling to reduce uncertainty. Bulletin of the American Meteorological Society, 98(9), 1857–1877, doi: 10.1175/bams-d-15-00317.1.
Reddington, C.L. et al., 2019: Exploring the impacts of anthropogenic emission sectors on PM2.5 and human health in South and East Asia. Atmospheric Chemistry and Physics, 19(18), 11887–11910, doi: 10.5194/acp-19-11887-2019.
Reis, S. et al., 2012: From acid rain to climate change. Science, 338(6111), 1153–1154, doi: 10.1126/science.1226514.
Ren, L. et al., 2020: Source attribution of Arctic black carbon and sulfate aerosols and associated Arctic surface warming during 1980–2018. Atmospheric Chemistry and Physics, 20(14), 9067–9085, doi: 10.5194/acp-20-9067-2020.
Riahi, K. et al., 2017: The Shared Socioeconomic Pathways and their energy, land use, and greenhouse gas emissions implications: An overview. Global Environmental Change, 42, 153–168, doi: 10.1016/j.gloenvcha.2016.05.009.
Ricciardelli, I. et al., 2017: A three-year investigation of daily PM2.5 main chemical components in four sites: the routine measurement program of the Supersito Project (Po Valley, Italy). Atmospheric Environment, 152, 418–430, doi: 10.1016/j.atmosenv.2016.12.052.
Riddick, S. et al., 2016: Estimate of changes in agricultural terrestrial nitrogen pathways and ammonia emissions from 1850 to present in the Community Earth System Model. Biogeosciences, 13(11), 3397–3426, doi: 10.5194/bg-13-3397-2016.
Rieder, H.E. et al., 2018: Combining model projections with site-level observations to estimate changes in distributions and seasonality of ozone in surface air over the U.S.A. Atmospheric Environment, 193, 302–315, doi: 10.1016/j.atmosenv.2018.07.042.
Rigby, M. et al., 2017: Role of atmospheric oxidation in recent methane growth. Proceedings of the National Academy of Sciences, 114(21), 5373–5377, doi: 10.1073/pnas.1616426114.
Robinson, M.A., M.R. Olson, Z.G. Liu, and J.J. Schauer, 2015: The effects of emission control strategies on light-absorbing carbon emissions from a modern heavy-duty diesel engine. Journal of the Air & Waste Management Association, 65(6), 759–766, doi: 10.1080/10962247.2015.1005850.
Robock, A., L. Oman, and G.L. Stenchikov, 2008: Regional climate responses to geoengineering with tropical and Arctic SO2 injections. Journal of Geophysical Research: Atmospheres, 113(D16), D16101, doi: 10.1029/2008jd010050.
Rogelj, J. et al., 2014a: Air-pollution emission ranges consistent with the representative concentration pathways. Nature Climate Change, 4(6), 446–450, doi: 10.1038/nclimate2178.
Rogelj, J. et al., 2014b: Disentangling the effects of CO2 and short-lived climate forcer mitigation. Proceedings of the National Academy of Sciences, 111(46), 16325–16330, doi: 10.1073/pnas.1415631111.
Rogelj, J. et al., 2017: Understanding the origin of Paris Agreement emission uncertainties. Nature Communications, 8(1), 1–12, doi: 10.1038/ncomms15748.
Rogelj, J. et al., 2018a: Scenarios towards limiting global mean temperature increase below 1.5°C. Nature Climate Change, 8(4), 325–332, doi: 10.1038/s41558-018-0091-3.
Rogelj, J. et al., 2018b: Mitigation Pathways Compatible With 1.5°C in the Context of Sustainable Development. In: Global Warming of 1.5°C. An IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty[Masson-Delmotte, V., P. Zhai, H.O. Pörtner, D. Roberts, J. Skea, P.R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J.B.R. Matthews, Y. Chen, X. Zhou, M.I. Gomis, E. Lonnoy, T. Maycock, M. Tignor, and T. Waterfield (eds.)]. In Press, pp. 93–174, www.ipcc.ch/sr15/chapter/chapter-2/.
Romer, P.S. et al., 2018: Effects of temperature-dependent NOx emissions on continental ozone production. Atmospheric Chemistry and Physics, 18(4), 2601–2614, doi: 10.5194/acp-18-2601-2018.
Rooney, B. et al., 2020: Air quality impact of the Northern California Camp Fire of November 2018. Atmospheric Chemistry and Physics, 20(23), 14597–14616, doi: 10.5194/acp-20-14597-2020.
Rosenstiel, T.N., M.J. Potosnak, K.L. Griffin, R. Fall, and R.K. Monson, 2003: Increased CO2 uncouples growth from isoprene emission in an agriforest ecosystem. Nature, 421(6920), 256–259, doi: 10.1038/nature01312.
Rossabi, S. and D. Helmig, 2018: Changes in Atmospheric Butanes and Pentanes and Their Isomeric Ratios in the Continental United States. Journal of Geophysical Research: Atmospheres, 123(7), 3772–3790, doi: 10.1002/2017jd027709.
Rowlinson, M.J. et al., 2019: Impact of El Niño-Southern Oscillation on the interannual variability of methane and tropospheric ozone. Atmospheric Chemistry and Physics, 19(13), 8669–8686, doi: 10.5194/acp-19-8669-2019.
Ru, M., D.T. Shindell, K.M. Seltzer, S. Tao, and Q. Zhong, 2018: The long-term relationship between emissions and economic growth for SO2, CO2, and BC. Environmental Research Letters, 13(12), 124021, doi: 10.1088/1748-9326/aaece2.
Ryder, C.L. et al., 2018: Coarse-mode mineral dust size distributions, composition and optical properties from AER-D aircraft measurements over the tropical eastern Atlantic. Atmospheric Chemistry and Physics, 18(23), 17225–17257, doi: 10.5194/acp-18-17225-2018.
Saari, R.K., N.E. Selin, S. Rausch, and T.M. Thompson, 2015: A self-consistent method to assess air quality co-benefits from U.S. climate policies. Journal of the Air & Waste Management Association, 65(1), 74–89, doi: 10.1080/10962247.2014.959139.
Sadiq, M., A.P.K. Tai, D. Lombardozzi, and M. Val Martin, 2017: Effects of ozone–vegetation coupling on surface ozone air quality via biogeochemical and meteorological feedbacks. Atmospheric Chemistry and Physics, 17(4), 3055–3066, doi: 10.5194/acp-17-3055-2017.
Saiz-Lopez, A. et al., 2012: Estimating the climate significance of halogen-driven ozone loss in the tropical marine troposphere. Atmospheric Chemistry and Physics, 12(9), 3939–3949, doi: 10.5194/acp-12-3939-2012.
Sajeev, E.P.M., B. Amon, C. Ammon, W. Zollitsch, and W. Winiwarter, 2018: Evaluating the potential of dietary crude protein manipulation in reducing ammonia emissions from cattle and pig manure: A meta-analysis. Nutrient Cycling in Agroecosystems, 110(1), 161–175, doi: 10.1007/s10705-017-9893-3.
Saliba, G. et al., 2019: Factors driving the seasonal and hourly variability of sea-spray aerosol number in the North Atlantic. Proceedings of the National Academy of Sciences, 116(41), 20309–20314, doi: 10.1073/pnas.1907574116.
Salter, M.E., E.D. Nilsson, A. Butcher, and M. Bilde, 2014: On the seawater temperature dependence of the sea spray aerosol generated by a continuous plunging jet. Journal of Geophysical Research: Atmospheres, 119(14), 9052–9072, doi: 10.1002/2013jd021376.
Samset, B.H., J.S. Fuglestvedt, and M.T. Lund, 2020: Delayed emergence of a global temperature response after emission mitigation. Nature Communications, 11(1), 3261, doi: 10.1038/s41467-020-17001-1.
Samset, B.H. et al., 2013: Black carbon vertical profiles strongly affect its radiative forcing uncertainty. Atmospheric Chemistry and Physics, 13(5), 2423–2434, doi: 10.5194/acp-13-2423-2013.
Samset, B.H. et al., 2016: Fast and slow precipitation responses to individual climate forcers: A PDRMIP multimodel study. Geophysical Research Letters, 43(6), 2782–2791, doi: 10.1002/2016gl068064.
Samset, B.H. et al., 2018: Climate Impacts From a Removal of Anthropogenic Aerosol Emissions. Geophysical Research Letters, 45(2), 1020–1029, doi: 10.1002/2017gl076079.
Samyn, D., C. Vega, H. Motoyama, and V. Pohjola, 2012: Nitrate and Sulfate Anthropogenic Trends in the 20th Century from Five Svalbard Ice Cores. Arctic, Antarctic, and Alpine Research, 44(4), 490–499, doi: 10.1657/1938-4246-44.4.490.
Sand, M., T.K. Berntsen, Seland, and J.E. Kristjánsson, 2013a: Arctic surface temperature change to emissions of black carbon within Arctic or midlatitudes. Journal of Geophysical Research: Atmospheres, 118(14), 7788–7798, doi: 10.1002/jgrd.50613.
Sand, M., T.K. Berntsen, A.M.L. Ekman, H.-C. Hansson, and A. Lewinschal, 2020: Surface temperature response to regional black carbon emissions: do location and magnitude matter?Atmospheric Chemistry and Physics, 20(5), 3079–3089, doi: 10.5194/acp-20-3079-2020.
Sand, M. et al., 2013b: The arctic response to remote and local forcing of black carbon. Atmospheric Chemistry and Physics, 13(1), 211–224, doi: 10.5194/acp-13-211-2013.
Sand, M. et al., 2016: Response of Arctic temperature to changes in emissions of short-lived climate forcers. Nature Climate Change, 6(3), 286–289, doi: 10.1038/nclimate2880.
Sandrini, S. et al., 2016: Size-resolved aerosol composition at an urban and a rural site in the Po Valley in summertime: implications for secondary aerosol formation. Atmospheric Chemistry and Physics, 16(17), 10879–10897, doi: 10.5194/acp-16-10879-2016.
Sarkar, C., A. Roy, A. Chatterjee, S.K. Ghosh, and S. Raha, 2019: Factors controlling the long-term (2009–2015) trend of PM 2.5 and black carbon aerosols at eastern Himalaya, India. Science of the Total Environment, 656, 280–296, doi: 10.1016/j.scitotenv.2018.11.367.
Sarwar, G. et al., 2015: Impact of Enhanced Ozone Deposition and Halogen Chemistry on Tropospheric Ozone over the Northern Hemisphere. Environmental Science & Technology, 49(15), 9203–9211, doi: 10.1021/acs.est.5b01657.
Saunois, M. et al., 2016: The global methane budget 2000–2012.Earth System Science Data, 8(2), 697–751, doi: 10.5194/essd-8-697-2016.
Saunois, M. et al., 2020: The Global Methane Budget 2000–2017. Earth System Science Data, 12(3), 1561–1623, doi: 10.5194/essd-12-1561-2020.
Schiferl, L.D. et al., 2016: Interannual variability of ammonia concentrations over the United States: Sources and implications. Atmospheric Chemistry and Physics, 16(18), 12305–12328, doi: 10.5194/acp-16-12305-2016.
Schmale, J., J. van Aardenne, and E. Von Schneidemesser, 2014a: New Directions: Support for integrated decision-making in air and climate policies – Development of a metrics-based information portal. Atmospheric Environment, 90, 146–148, doi: 10.1016/j.atmosenv.2014.03.016.
Schmale, J., D. Shindell, E. Von Schneidemesser, I. Chabay, and M. Lawrence, 2014b: Air pollution: Clean up our skies. Nature, 515(7527), 335–337, doi: 10.1038/515335a.
Schneider, P., W.A. Lahoz, and R. Van Der A, 2015: Recent satellite-based trends of tropospheric nitrogen dioxide over large urban agglomerations worldwide. Atmospheric Chemistry and Physics, 15(3), 1205–1220, doi: 10.5194/acp-15-1205-2015.
Schnell, J.L. and M.J. Prather, 2017: Co-occurrence of extremes in surface ozone, particulate matter, and temperature over eastern North America. Proceedings of the National Academy of Sciences, 114(11), 2854–2859, doi: 10.1073/pnas.1614453114.
Schnell, J.L. et al., 2016: Effect of climate change on surface ozone over North America, Europe, and East Asia. Geophysical Research Letters, 43(7), 3509–3518, doi: 10.1002/2016gl068060.
Schnell, J.L. et al., 2018: Exploring the relationship between surface PM2.5 and meteorology in Northern India. Atmospheric Chemistry and Physics, 18(14), 10157–10175, doi: 10.5194/acp-18-10157-2018.
Schroder, J.C. et al., 2015: Size-resolved observations of refractory black carbon particles in cloud droplets at a marine boundary layer site. Atmospheric Chemistry and Physics, 15(3), 1367–1383, doi: 10.5194/acp-15-1367-2015.
Schultz, M.G. et al., 2015: The Global Atmosphere Watch reactive gases measurement network. Elementa: Science of the Anthropocene, 3, 000067, doi: 10.12952/journal.elementa.000067.
Schulz, H. et al., 2019: High Arctic aircraft measurements characterising black carbon vertical variability in spring and summer. Atmospheric Chemistry and Physics, 19(4), 2361–2384, doi: 10.5194/acp-19-2361-2019.
Schwarber, A.K., S.J. Smith, C.A. Hartin, B.A. Vega-Westhoff, and R. Sriver, 2019: Evaluating climate emulation: fundamental impulse testing of simple climate models. Earth System Dynamics, 10(4), 729–739, doi: 10.5194/esd-10-729-2019.
Schwarz, J.P. et al., 2013: Global-scale seasonally resolved black carbon vertical profiles over the Pacific. Geophysical Research Letters, 40(20), 5542–5547, doi: 10.1002/2013gl057775.
Schwinger, J. et al., 2017: Amplification of global warming through pH dependence of DMS production simulated with a fully coupled Earth system model. Biogeosciences, 14(15), 3633–3648, doi: 10.5194/bg-14-3633-2017.
Scott, C.E. et al., 2017: Impact on short-lived climate forcers (SLCFs) from a realistic land-use change scenario: Via changes in biogenic emissions. Faraday Discussions, 200, 101–120, doi: 10.1039/c7fd00028f .
Scott, C.E. et al., 2018: Substantial large-scale feedbacks between natural aerosols and climate. Nature Geoscience, 11(1), 44–48, doi: 10.1038/s41561-017-0020-5.
Sekiya, T. and K. Sudo, 2014: Roles of transport and chemistry processes in global ozone change on interannual and multidecadal time scales. Journal of Geophysical Research: Atmospheres, 119(8), 4903–4921, doi: 10.1002/2013jd020838.
Serrano, H.C. et al., 2019: Measuring and mapping the effectiveness of the European Air Quality Directive in reducing N and S deposition at the ecosystem level. Science of the Total Environment, 647, 1531–1538, doi: 10.1016/j.scitotenv.2018.08.059.
Shah, N., M. Wei, V. Letschert, and A. Phadke, 2015: Benefits of Leapfrogging to Superefficiency and Low Global Warming Potential Refrigerants in Room Air Conditioning. Report No. LBNL-1003671, Ernest Orlando Lawrence Berkeley National Laboratory (LBNL), Berkeley, CA, USA, 39 pp., doi: 10.2172/1397235.
Sharma, S. et al., 2013: 16-year simulation of Arctic black carbon: Transport, source contribution, and sensitivity analysis on deposition. Journal of Geophysical Research: Atmospheres, 118(2), 943–964, doi: 10.1029/2012jd017774.
Sharma, S. et al., 2017: An evaluation of three methods for measuring black carbon in Alert, Canada. Atmospheric Chemistry and Physics, 17(24), 15225–15243, doi: 10.5194/acp-17-15225-2017.
Shen, L., L.J. Mickley, and L.T. Murray, 2017: Influence of 2000–2050 climate change on particulate matter in the United States: Results from a new statistical model. Atmospheric Chemistry and Physics, 17(6), 4355–4367, doi: 10.5194/acp-17-4355-2017.
Shen, L. et al., 2019: The 2005–2016 Trends of Formaldehyde Columns Over China Observed by Satellites: Increasing Anthropogenic Emissions of Volatile Organic Compounds and Decreasing Agricultural Fire Emissions. Geophysical Research Letters, 46(8), 4468–4475, doi: 10.1029/2019gl082172.
Shephard, M.W. and K.E. Cady-Pereira, 2015: Cross-track Infrared Sounder (CrIS) satellite observations of tropospheric ammonia. Atmospheric Measurement Techniques, 8(3), 1323–1336, doi: 10.5194/amt-8-1323-2015.
Sheppard, L.J. et al., 2011: Dry deposition of ammonia gas drives species change faster than wet deposition of ammonium ions: evidence from a long-term field manipulation. Global Change Biology, 17(12), 3589–3607, doi: 10.1111/j.1365-2486.2011.02478.x.
Sherwen, T. et al., 2016: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem. Atmospheric Chemistry and Physics, 16(18), 12239–12271, doi: 10.5194/acp-16-12239-2016.
Sherwood, S.C. et al., 2020: An Assessment of Earth’s Climate Sensitivity Using Multiple Lines of Evidence. Reviews of Geophysics, 58(4), e2019RG000678, doi: 10.1029/2019rg000678.
Shi, G. et al., 2017: pH of Aerosols in a Polluted Atmosphere: Source Contributions to Highly Acidic Aerosol. Environmental Science & Technology,51(8), 4289–4296, doi: 10.1021/acs.est.6b05736.
Shi, Z. et al., 2021: Abrupt but smaller than expected changes in surface air quality attributable to COVID-19 lockdowns. Science Advances, 7(3), eabd6696, doi: 10.1126/sciadv.abd6696.
Shindell, D.T. and G. Faluvegi, 2009: Climate response to regional radiative forcing during the twentieth century. Nature Geoscience, 2(4), 294–300, doi: 10.1038/ngeo473.
Shindell, D.T. and C.J. Smith, 2019: Climate and air-quality benefits of a realistic phase-out of fossil fuels. Nature, 573(7774), 408–411, doi: 10.1038/s41586-019-1554-z.
Shindell, D.T., Y. Lee, and G. Faluvegi, 2016: Climate and health impacts of US emissions reductions consistent with 2°C. Nature Climate Change, 6(5), 503–507, doi: 10.1038/nclimate2935.
Shindell, D.T., J.S. Fuglestvedt, and W.J. Collins, 2017a: The social cost of methane: theory and applications. Faraday Discussions, 200, 429–451, doi: 10.1039/c7fd00009j.
Shindell, D.T., G. Faluvegi, L. Rotstayn, and G. Milly, 2015: Spatial patterns of radiative forcing and surface temperature response. Journal of Geophysical Research: Atmospheres, 120(11), 5385–5403, doi: 10.1002/2014jd022752.
Shindell, D.T., G. Faluvegi, K. Seltzer, and C. Shindell, 2018: Quantified, localized health benefits of accelerated carbon dioxide emissions reductions. Nature Climate Change, 8(4), 1–5, doi: 10.1038/s41558-018-0108-y.
Shindell, D.T. et al., 2009: Improved Attribution of Climate Forcing to Emissions. Science, 326(5953), 716–718, doi: 10.1126/science.1174760.
Shindell, D.T. et al., 2012: Simultaneously mitigating near-term climate change and improving human health and food security. Science, 335(6065), 183–189, doi: 10.1126/science.1210026.
Shindell, D.T. et al., 2013: Radiative forcing in the ACCMIP historical and future climate simulations. Atmospheric Chemistry and Physics, 13(6), 2939–2974, doi: 10.5194/acp-13-2939-2013.
Shindell, D.T. et al., 2017b: A climate policy pathway for near- and long-term benefits. Science, 356(6337), 493–494, doi: 10.1126/science.aak9521.
Shoemaker, J.K., D.P. Schrag, M.J. Molina, and V. Ramanathan, 2013: What role for short-lived climate pollutants in mitigation policy?Science, 342(6164), 1323–1324, doi: 10.1126/science.1240162.
Shrivastava, M. et al., 2017: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Reviews of Geophysics, 55(2), 509–559, doi: 10.1002/2016rg000540.
Sicard, P. et al., 2020: Amplified ozone pollution in cities during the COVID-19 lockdown. Science of The Total Environment, 735, 139542, doi: 10.1016/j.scitotenv.2020.139542.
Sickles II, J.E. and D.S. Shadwick, 2015: Air quality and atmospheric deposition in the eastern US: 20 years of change. Atmospheric Chemistry and Physics, 15(1), 173–197, doi: 10.5194/acp-15-173-2015.
Silva, R.A., Z. Adelman, M.M. Fry, and J.J. West, 2016: The Impact of Individual Anthropogenic Emissions Sectors on the Global Burden of Human Mortality due to Ambient Air Pollution. Environmental Health Perspectives, 124(11), 1776–1784, doi: 10.1289/ehp177.
Silver, B., C.L. Reddington, S.R. Arnold, and D. Spracklen, 2018: Substantial changes in air pollution across China during 2015–2017. Environmental Research Letters, 13(11), 114012, doi: 10.1088/1748-9326/aae718.
Silvern, R.F. et al., 2018: Observed NO/NO2 Ratios in the Upper Troposphere Imply Errors in NO-NO2-O3 Cycling Kinetics or an Unaccounted NOx Reservoir. Geophysical Research Letters, 45(9), 4466–4474, doi: 10.1029/2018gl077728.
Simpson, I.J. et al., 2012: Long-term decline of global atmospheric ethane concentrations and implications for methane. Nature, 488(7412), 490–494, doi: 10.1038/nature11342.
Singh, V., K. Ravindra, L. Sahu, and R. Sokhi, 2018: Trends of atmospheric black carbon concentration over United Kingdom. Atmospheric Environment, 178, 148–157, doi: 10.1016/j.atmosenv.2018.01.030.
Sitch, S., P.M. Cox, W.J. Collins, and C. Huntingford, 2007: Indirect radiative forcing of climate change through ozone effects on the land-carbon sink. Nature, 448(7155), 791–794, doi: 10.1038/nature06059.
Six, K.D. et al., 2013: Global warming amplified by reduced sulphur fluxes as a result of ocean acidification. Nature Climate Change, 3(11), 975–978, doi: 10.1038/nclimate1981.
Skowron, A., D.S. Lee, R.R. De León, L.L. Lim, and B. Owen, 2021: Greater fuel efficiency is potentially preferable to reducing NOx emissions for aviation’s climate impacts. Nature Communications, 12(1), 564, doi: 10.1038/s41467-020-20771-3.
Smith, C.J. et al., 2018: FAIR v1.3: a simple emissions-based impulse response and carbon cycle model. Geoscientific Model Development, 11(6), 2273–2297, doi: 10.5194/gmd-11-2273-2018.
Smith, C.J. et al., 2019: Current fossil fuel infrastructure does not yet commit us to 1.5°C warming. Nature Communications, 10(1), 101, doi: 10.1038/s41467-018-07999-w.
Smith, S.J. and A. Mizrahi, 2013: Near-term climate mitigation by short-lived forcers. Proceedings of the National Academy of Sciences, 110(35), 14202–14206, doi: 10.1073/pnas.1308470110.
Smith, S.J. et al., 2020: Impact of methane and black carbon mitigation on forcing and temperature: a multi-model scenario analysis. Climatic Change, 163(3), 1427–1442, doi: 10.1007/s10584-020-02794-3.
Snider, G. et al., 2015: SPARTAN: a global network to evaluate and enhance satellite-based estimates of ground-level particulate matter for global health applications. Atmospheric Measurement Techniques, 8(1), 505–521, doi: 10.5194/amt-8-505-2015.
Snider, G. et al., 2016: Variation in global chemical composition of PM2.5: emerging results from SPARTAN. Atmospheric Chemistry and Physics, 16(15), 9629–9653, doi: 10.5194/acp-16-9629-2016.
Soares, J. et al., 2016: Impact of Climate Change on the Production and Transport of Sea Salt Aerosol on European Seas. In: Air Pollution Modeling and its Application XXIV[Steyn, D. and N. Chaumerliac (eds.)]. Springer Proceedings in Complexity, Springer, Cham, Switzerland, pp. 207–212, doi: 10.1007/978-3-319-24478-5_34.
Sofiev, M. et al., 2018: Cleaner fuels for ships provide public health benefits with climate tradeoffs. Nature Communications, 9(1), 406, doi: 10.1038/s41467-017-02774-9.
Solomon, P.A. et al., 2014: U.S. National PM2.5 Chemical Speciation Monitoring Networks – CSN and IMPROVE: Description of networks. Journal of the Air & Waste Management Association, 64(12), 1410–1438, doi: 10.1080/10962247.2014.956904.
Spracklen, D., S.R. Arnold, L. Conibear, E.W. Butt, and C. Knote, 2018: Residential energy use emissions dominate health impacts from exposure to ambient particulate matter in India. Nature Communications, 9(1), 617, doi: 10.1038/s41467-018-02986-7.
Squire, O.J. et al., 2015: Influence of isoprene chemical mechanism on modelled changes in tropospheric ozone due to climate and land use over the 21st century. Atmospheric Chemistry and Physics, 15(9), 5123–5143, doi: 10.5194/acp-15-5123-2015.
Stanelle, T., I. Bey, T. Raddatz, C. Reick, and I. Tegen, 2014: Anthropogenically induced changes in twentieth century mineral dust burden and the associated impact on radiative forcing. Journal of Geophysical Research: Atmospheres, 119(23), 13526–13546, doi: 10.1002/2014jd022062.
Stavrakou, T. et al., 2014: Isoprene emissions over Asia 1979–2012: impact of climate and land-use changes. Atmospheric Chemistry and Physics, 14(9), 4587–4605, doi: 10.5194/acp-14-4587-2014.
Stechow, C. et al., 2016: 2°C and SDGs: United they stand, divided they fall?Environmental Research Letters, 11, 34022, doi: 10.1088/1748-9326/11/3/034022.
Stein, O. et al., 2014: On the wintertime low bias of Northern Hemisphere carbon monoxide found in global model simulations. Atmospheric Chemistry and Physics, 14(17), 9295–9316, doi: 10.5194/acp-14-9295-2014.
Stevens, B. et al., 2017: MACv2-SP: a parameterization of anthropogenic aerosol optical properties and an associated Twomey effect for use in CMIP6. Geoscientific Model Development, 10(1), 433–452, doi: 10.5194/gmd-10-433-2017.
Stevenson, D.S. et al., 2006: Multimodel ensemble simulations of present-day and near-future tropospheric ozone. Journal of Geophysical Research: Atmospheres, 111(D8), D08301, doi: 10.1029/2005jd006338.
Stevenson, D.S. et al., 2013: Tropospheric ozone changes, radiative forcing and attribution to emissions in the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmospheric Chemistry and Physics, 13(6), 3063–3085, doi: 10.5194/acp-13-3063-2013.
Stevenson, D.S. et al., 2020: Trends in global tropospheric hydroxyl radical and methane lifetime since 1850 from AerChemMIP. Atmospheric Chemistry and Physics, 20(21), 12905–12920, doi: 10.5194/acp-20-12905-2020.
Stjern, C.W. et al., 2017: Rapid Adjustments Cause Weak Surface Temperature Response to Increased Black Carbon Concentrations. Journal of Geophysical Research: Atmospheres, 122(21), 11462–11481, doi: 10.1002/2017jd027326.
Stockwell, W.R., E. Saunders, W.S. Goliff, and R.M. Fitzgerald, 2020: A perspective on the development of gas-phase chemical mechanisms for Eulerian air quality models. Journal of the Air & Waste Management Association, 70(1), 44–70, doi: 10.1080/10962247.2019.1694605.
Stohl, A. et al., 2013: Black carbon in the Arctic: the underestimated role of gas flaring and residential combustion emissions. Atmospheric Chemistry and Physics, 13(17), 8833–8855, doi: 10.5194/acp-13-8833-2013.
Stohl, A. et al., 2015: Evaluating the climate and air quality impacts of short-lived pollutants. Atmospheric Chemistry and Physics, 15(18), 10529–10566, doi: 10.5194/acp-15-10529-2015.
Storelvmo, T. et al., 2013: Cirrus cloud seeding has potential to cool climate. Geophysical Research Letters, 40(1), 178–182, doi: 10.1029/2012gl054201.
Strada, S. and N. Unger, 2016: Potential sensitivity of photosynthesis and isoprene emission to direct radiative effects of atmospheric aerosol pollution. Atmospheric Chemistry and Physics, 16(7), 4213–4234, doi: 10.5194/acp-16-4213-2016.
Strefler, J., G. Luderer, E. Kriegler, and M. Meinshausen, 2014: Can air pollutant controls change global warming?Environmental Science & Policy, 41, 33–43, doi: 10.1016/j.envsci.2014.04.009.
Strode, S.A. et al., 2015: Implications of carbon monoxide bias for methane lifetime and atmospheric composition in chemistry climate models. Atmospheric Chemistry and Physics, 15(20), 11789–11805, doi: 10.5194/acp-15-11789-2015.
Strode, S.A. et al., 2016: Interpreting space-based trends in carbon monoxide with multiple models. Atmospheric Chemistry and Physics, 16(11), 7285–7294, doi: 10.5194/acp-16-7285-2016.
Struthers, H. et al., 2013: Climate-induced changes in sea salt aerosol number emissions: 1870 to 2100. Journal of Geophysical Research: Atmospheres, 118(2), 670–682, doi: 10.1002/jgrd.50129.
Stuber, N., M. Ponater, and R. Sausen, 2001: Is the climate sensitivity to ozone perturbations enhanced by stratospheric water vapor feedback?Geophysical Research Letters, 28(15), 2887–2890, doi: 10.1029/2001gl013000.
Sun, G.E. et al., 2012: Interactive influences of ozone and climate on streamflow of forested watersheds. Global Change Biology, 18(11), 3395–3409, doi: 10.1111/j.1365-2486.2012.02787.x.
Sun, W., P. Hess, and C. Liu, 2017: The impact of meteorological persistence on the distribution and extremes of ozone. Geophysical Research Letters, 44(3), 1545–1553, doi: 10.1002/2016gl071731.
Sun, W. et al., 2018: Long-Term Trends of Anthropogenic SO2, NOx, CO, and NMVOCs Emissions in China. Earth’s Future, 6(8), 1112–1133, doi: 10.1029/2018ef000822.
Sutton, M.A. et al., 2013: Towards a climate-dependent paradigm of ammonia emission and deposition. Philosophical Transactions of the Royal Society B: Biological Sciences, 368(1621), 20130166, doi: 10.1098/rstb.2013.0166.
Szogs, S. et al., 2017: Impact of LULCC on the emission of BVOCs during the 21st century. Atmospheric Environment, 165, 73–87, doi: 10.1016/j.atmosenv.2017.06.025.
Tan, J. et al., 2018: Multi-model study of HTAP II on sulfur and nitrogen deposition. Atmospheric Chemistry and Physics, 18(9), 6847–6866, doi: 10.5194/acp-18-6847-2018.
Tanaka, K., M.T. Lund, B. Aamaas, and T. Berntsen, 2018: Climate effects of non-compliant Volkswagen diesel cars. Environmental Research Letters, 13(4), 44020, doi: 10.1088/1748-9326/aab18c.
Tang, Y.S. et al., 2018: Drivers for spatial, temporal and long-term trends in atmospheric ammonia and ammonium in the UK. Atmospheric Chemistry and Physics, 18(2), 705–733, doi: 10.5194/acp-18-705-2018.
Tanimoto, H., K. Ikeda, K.F. Boersma, R.J. A, and S. Garivait, 2015: Interannual variability of nitrogen oxides emissions from boreal fires in Siberia and Alaska during 1996–2011 as observed from space. Environmental Research Letters, 10(6), 065004, doi: 10.1088/1748-9326/10/6/065004.
Taylor, J.W. et al., 2014: Size-dependent wet removal of black carbon in Canadian biomass burning plumes. Atmospheric Chemistry and Physics, 14(24), 13755–13771, doi: 10.5194/acp-14-13755-2014.
Té, Y. et al., 2016: Seasonal variability of surface and column carbon monoxide over the megacity Paris, high-altitude Jungfraujoch and Southern Hemispheric Wollongong stations. Atmospheric Chemistry and Physics, 16(17), 10911–10925, doi: 10.5194/acp-16-10911-2016.
Theobald, M.R. et al., 2019: An evaluation of European nitrogen and sulfur wet deposition and their trends estimated by six chemistry transport models for the period 1990–2010. Atmospheric Chemistry and Physics, 19(1), 379–405, doi: 10.5194/acp-19-379-2019.
Thompson, T.M., S. Rausch, R.K. Saari, and N.E. Selin, 2016: Air quality co-benefits of subnational carbon policies. Journal of the Air & Waste Management Association, 66(10), 988–1002, doi: 10.1080/10962247.2016.1192071.
Thornhill, G.D. et al., 2021a: Climate-driven chemistry and aerosol feedbacks in CMIP6 Earth system models. Atmospheric Chemistry and Physics, 21(2), 1105–1126, doi: 10.5194/acp-21-1105-2021.
Thornhill, G.D. et al., 2021b: Effective radiative forcing from emissions of reactive gases and aerosols – a multi-model comparison. Atmospheric Chemistry and Physics, 21(2), 853–874, doi: 10.5194/acp-21-853-2021.
Tilmes, S. et al., 2019: Climate Forcing and Trends of Organic Aerosols in the Community Earth System Model (CESM2). Journal of Advances in Modeling Earth Systems, 11(12), 4323–4351, doi: 10.1029/2019ms001827.
Tohjima, Y. et al., 2020: Detection of fossil-fuel CO2 plummet in China due to COVID-19 by observation at Hateruma. Scientific Reports, 10(1), 18688, doi: 10.1038/s41598-020-75763-6.
Tong, D. et al., 2020: Dynamic projection of anthropogenic emissions in China: methodology and 2015–2050 emission pathways under a range of socioeconomic, climate policy, and pollution control scenarios. Atmospheric Chemistry and Physics, 20(9), 5729–5757, doi: 10.5194/acp-20-5729-2020.
Tsigaridis, K. et al., 2014: The AeroCom evaluation and intercomparison of organic aerosol in global models. Atmospheric Chemistry and Physics, 14(19), 10845–10895, doi: 10.5194/acp-14-10845-2014.
Turner, A.J., C. Frankenberg, P.O. Wennberg, and D.J. Jacob, 2017: Ambiguity in the causes for decadal trends in atmospheric methane and hydroxyl. Proceedings of the National Academy of Sciences, 114(21), 5367–5372, doi: 10.1073/pnas.1616020114.
Turner, A.J., I. Fung, V. Naik, L.W. Horowitz, and R.C. Cohen, 2018: Modulation of hydroxyl variability by ENSO in the absence of external forcing. Proceedings of the National Academy of Sciences, 115(36), 8931–8936, doi: 10.1073/pnas.1807532115.
Turnock, S.T. et al., 2016: The impact of European legislative and technology measures to reduce air pollutants on air quality, human health and climate. Environmental Research Letters, 11(2), 024010, doi: 10.1088/1748-9326/11/2/024010.
Turnock, S.T. et al., 2019: The Impact of Changes in Cloud Water pH on Aerosol Radiative Forcing. Geophysical Research Letters, 46(7), 4039–4048, doi: 10.1029/2019gl082067.
Turnock, S.T. et al., 2020: Historical and future changes in air pollutants from CMIP6 models. Atmospheric Chemistry and Physics, 20(23), 14547–14579, doi: 10.5194/acp-20-14547-2020.
Twomey, S., 1977: The Influence of Pollution on the Shortwave Albedo of Clouds. Journal of the Atmospheric Sciences, 34(7), 1149–1152, doi: 10.1175/1520-0469(1977)034<1149:tiopot>2.0.co;2.
Tzompa-Sosa, Z.A. et al., 2019: Atmospheric Implications of Large C2-C5Alkane Emissions From the U.S. Oil and Gas Industry. Journal of Geophysical Research: Atmospheres, 124(2), 1148–1169, doi: 10.1029/2018jd028955.
Uherek, E. et al., 2010: Transport impacts on atmosphere and climate: Land transport. Atmospheric Environment, 44(37), 4772–4816, https://treaties.un.org/doc/publication/unts/volume 1522/volume-1522-i-26369-english.pdf
UN, 1989: The Montreal Protocol on Substances that Deplete the Ozone Layer. In: United Nations – Treaty Series No. 26369. United Nations (UN), New York, NY, USA, pp. 29–111, doi: treaties.un.org/doc/publication/unts/volume%201522/volume-1522-i-26369-english.pdf .
UNECE, 2010: Hemispheric Transport of Air Pollution. Part A: Ozone and Particulate Matter[Dentener, F.J., T.J. Keating, and H. Akimoto (eds.)]. Air Pollution Studies No. 17, United Nations Economic Commission for Europe (UNECE). United Nations (UN), New York, NY, USA and Geneva, Switzerland, 278 pp., https://unece.org/hemispheric-transport-air-pollution.
UNEP, 2016: Amendment to the Montreal Protocol on Substances that Deplete the Ozone Layer. In: Report of theTwenty-EighthMeeting of the Parties to the Montreal Protocol on Substances that Deplete the Ozone Layer. UNEP/OzL.Pro.28/12, United Nations Environment Programme (UNEP), Nairobi, Kenya, pp. 46–53, https://undocs.org/pdf?symbol=en/UNEP/OzL.Pro.28/12.
UNEP, 2019: Air Pollution in Asia and the Pacific: Science-BasedSolutions. United Nations Environment Program (UNEP), Nairobi, Kenya, 232 pp., www.ccacoalition.org/en/resources/air-pollution-asia-and-pacific-science-based-solutions-summary-full-report .
UNEP and WMO, 2011: Integrated Assessment of Black Carbon and Tropospheric Ozone. Summary for Decision Makers. UNEP/GC/26/INF/20, United Nations Environment Programme (UNEP) and World Meteorological Organization (WMO). UNEP, Nairobi, Kenya, 30 pp., http://hdl.handle.net/20.500.11822/8028.
UNEP and CCAC, 2018: Integrated Assessment of Short-lived Climate Pollutants in Latin America and the Caribbean: improving air quality while mitigating climate change. United Nations Environment Programme (UNEP) and Climate and Clean Air Coalition (CCAC). UNEP, Nairobi, Kenya, 101 pp., www.ccacoalition.org/en/resources/integrated-assessment-short-lived-climate-pollutants-latin-america-and-caribbean.
Unger, N., 2013: Isoprene emission variability through the twentieth century. Journal of Geophysical Research: Atmospheres, 118(24), 13606–13613, doi: 10.1002/2013jd020978.
Unger, N., 2014: Human land-use-driven reduction of forest volatiles cools global climate. Nature Climate Change, 4(10), 907–910, doi: 10.1038/nclimate2347.
Unger, N., X. Yue, and K.L. Harper, 2017: Aerosol climate change effects on land ecosystem services. Faraday Discussions, 200, 121–142, doi: 10.1039/c7fd00033b.
Unger, N., Y. Zheng, X. Yue, and K.L. Harper, 2020: Mitigation of ozone damage to the world’s land ecosystems by source sector. Nature Climate Change, 10(2), 134–137, doi: 10.1038/s41558-019-0678-3.
Unger, N. et al., 2010: Attribution of climate forcing to economic sectors. Proceedings of the National Academy of Sciences, 107(8), 3382–3387, doi: 10.1073/pnas.0906548107.
Unger, N. et al., 2013: Photosynthesis-dependent isoprene emission from leaf to planet in a global carbon–chemistry–climate model. Atmospheric Chemistry and Physics, 13(20), 10243–10269, doi: 10.5194/acp-13-10243-2013.
Val Martin, M. et al., 2015: How emissions, climate, and land use change will impact mid-century air quality over the United States: a focus on effects at national parks. Atmospheric Chemistry and Physics, 15(5), 2805–2823, doi: 10.5194/acp-15-2805-2015.
Van Damme, M. et al., 2015: Worldwide spatiotemporal atmospheric ammonia (NH3) columns variability revealed by satellite. Geophysical Research Letters, 42(20), 8660–8668, doi: 10.1002/2015gl065496.
Van Damme, M. et al., 2018: Industrial and agricultural ammonia point sources exposed. Nature, 564(7734), 99–103, doi: 10.1038/s41586- 018-0747-1.
Van Dingenen, R. et al., 2009: The global impact of ozone on agricultural crop yields under current and future air quality legislation. Atmospheric Environment, 43(3), 604–618, doi: 10.1016/j.atmosenv.2008.10.033.
Van Dingenen, R. et al., 2018: TM5-FASST: a global atmospheric source-receptor model for rapid impact analysis of emission changes on air quality and short-lived climate pollutants. Atmospheric Chemistry and Physics, 18(21), 16173–16211, doi: 10.5194/acp-18-16173-2018.
van Heerwaarden, C.C. et al., 2021: Record high solar irradiance in Western Europe during first COVID-19 lockdown largely due to unusual weather. Communications Earth & Environment, 2(1), 37, doi: 10.1038/s43247- 021-00110-0.
van Marle, M.J.E. et al., 2017: Historic global biomass burning emissions for CMIP6 (BB4CMIP) based on merging satellite observations with proxies and fire models (1750–2015). Geoscientific Model Development, 10(9), 3329–3357, doi: 10.5194/gmd-10-3329-2017.
van Vuuren, D.P. et al., 2011: The representative concentration pathways: an overview. Climatic Change, 109(1), 5–31, doi: 10.1007/s10584- 011-0148-z.
van Zanten, M.C., R.J. Wichink Kruit, R. Hoogerbrugge, E. Van der Swaluw, and W.A.J. van Pul, 2017: Trends in ammonia measurements in the Netherlands over the period 1993–2014. Atmospheric Environment, 148, 352–360, doi: 10.1016/j.atmosenv.2016.11.007.
Vandyck, T. et al., 2018: Air quality co-benefits for human health and agriculture counterbalance costs to meet Paris Agreement pledges. Nature Communications, 9(1), 4939, doi: 10.1038/s41467-018-06885-9.
VanLoocke, A., A.M. Betzelberger, E.A. Ainsworth, and C.J. Bernacchi, 2012: Rising ozone concentrations decrease soybean evapotranspiration and water use efficiency whilst increasing canopy temperature. New Phytologist, 195(1), 164–171, doi: 10.1111/j.1469-8137.2012.04152.x.
Vattioni, S. et al., 2019: Exploring accumulation-mode H2 SO4versus SO2 stratospheric sulfate geoengineering in a sectional aerosol–chemistry–climate model. Atmospheric Chemistry and Physics, 19(7), 4877–4897, doi: 10.5194/acp-19-4877-2019.
Veira, A., G. Lasslop, and S. Kloster, 2016: Wildfires in a warmer climate: Emission fluxes, emission heights, and black carbon concentrations in 2090–2099. Journal of Geophysical Research: Atmospheres, 121(7), 3195–3223, doi: 10.1002/2015jd024142.
Velders, G.J.M., D.W. Fahey, J.S. Daniel, M. McFarland, and S.O. Andersen, 2009: The large contribution of projected HFC emissions to future climate forcing. Proceedings of the National Academy of Sciences, 106(27), 10949–10954, doi: 10.1073/pnas.0902817106.
Velders, G.J.M., D.W. Fahey, J.S. Daniel, S.O. Andersen, and M. McFarland, 2015: Future atmospheric abundances and climate forcings from scenarios of global and regional hydrofluorocarbon (HFC) emissions. Atmospheric Environment, 123, 200–209, doi: 10.1016/j.atmosenv.2015.10.071.
Venkataraman, C. et al., 2018: Source influence on emission pathways and ambient PM2.5 pollution over India (2015–2050). Atmospheric Chemistry and Physics, 18(11), 8017–8039, doi: 10.5194/acp-18-8017-2018.
Venter, Z.S., K. Aunan, S. Chowdhury, and J. Lelieveld, 2020: COVID-19 lockdowns cause global air pollution declines. Proceedings of the National Academy of Sciences, 117(32), 18984–18990, doi: 10.1073/pnas.2006853117.
Vereecken, L., D.R. Glowacki, and M.J. Pilling, 2015: Theoretical Chemical Kinetics in Tropospheric Chemistry: Methodologies and Applications. Chemical Reviews, 115(10), 4063–4114, doi: 10.1021/cr500488p.
Vernier, J.-P. et al., 2018: BATAL: The Balloon Measurement Campaigns of the Asian Tropopause Aerosol Layer. Bulletin of the American Meteorological Society, 99(5), 955–973, doi: 10.1175/bams-d-17-0014.1.
Vet, R. et al., 2014: A global assessment of precipitation chemistry and deposition of sulfur, nitrogen, sea salt, base cations, organic acids, acidity and pH, and phosphorus. Atmospheric Environment, 93, 3–100, doi: 10.1016/j.atmosenv.2013.10.060.
Victor, D.G., D. Zaelke, and V. Ramanathan, 2015: Soot and short-lived pollutants provide political opportunity. Nature Climate Change, 5(9), 796–798, doi: 10.1038/nclimate2703.
Vignesh, P.P. et al., 2020: Assessment of CMIP6 Cloud Fraction and Comparison with Satellite Observations. Earth and Space Science, 7(2), e2019EA000975, doi: 10.1029/2019ea000975.
Vira, J., P. Hess, J. Melkonian, and W.R. Wieder, 2020: An improved mechanistic model for ammonia volatilization in Earth system models: Flow of Agricultural Nitrogen version 2 (FANv2). Geoscientific Model Development, 13(9), 4459–4490, doi: 10.5194/gmd-13-4459-2020.
Virts, K.S., J.M. Wallace, M.L. Hutchins, and R.H. Holzworth, 2013: Highlights of a New Ground-Based, Hourly Global Lightning Climatology. Bulletin of the American Meteorological Society, 94(9), 1381–1391, doi: 10.1175/bams-d-12-00082.1.
Visioni, D. et al., 2019: Seasonal Injection Strategies for Stratospheric Aerosol Geoengineering. Geophysical Research Letters, 46(13), 7790–7799, doi: 10.1029/2019gl083680.
von Schneidemesser, E. et al., 2015: Chemistry and the Linkages between Air Quality and Climate Change. Chemical Reviews, 115(10), 3856–3897, doi: 10.1021/acs.chemrev.5b00089.
Voulgarakis, A. et al., 2013: Analysis of present day and future OH and methane lifetime in the ACCMIP simulations. Atmospheric Chemistry and Physics, 13(5), 2563–2587, doi: 10.5194/acp-13-2563-2013.
Wang, B., J. Shuman, H.H. Shugart, and M.T. Lerdau, 2018: Biodiversity matters in feedbacks between climate change and air quality: a study using an individual-based model. Ecological Applications, 28(5), 1223–1231, doi: 10.1002/eap.1721.
Wang, H., P.J. Rasch, and G. Feingold, 2011: Manipulating marine stratocumulus cloud amount and albedo: a process-modelling study of aerosol–cloud–precipitation interactions in response to injection of cloud condensation nuclei. Atmospheric Chemistry and Physics, 11(9), 4237–4249, doi: 10.5194/acp-11-4237-2011.
Wang, M. et al., 2015: Trends of non-methane hydrocarbons (NMHC) emissions in Beijing during 2002-2013. Atmospheric Chemistry and Physics, 15(3), 1489–1502, doi: 10.5194/acp-15-1489-2015.
Wang, P., K. Chen, S. Zhu, P. Wang, and H. Zhang, 2020: Severe air pollution events not avoided by reduced anthropogenic activities during COVID-19 outbreak. Resources, Conservation and Recycling, 158, 104814, doi: 10.1016/j.resconrec.2020.104814.
Wang, Q. et al., 2014: Global budget and radiative forcing of black carbon aerosol: Constraints from pole-to-pole (HIPPO) observations across the Pacific. Journal of Geophysical Research: Atmospheres, 119(1), 195–206, doi: 10.1002/2013jd020824.
Wang, R. et al., 2014: Trend in Global Black Carbon Emissions from 1960 to 2007. Environmental Science & Technology, 48(12), 6780–6787, doi: 10.1021/es5021422.
Wang, S., M.E. Maltrud, S.M. Burrows, S.M. Elliott, and P. Cameron-Smith, 2018: Impacts of Shifts in Phytoplankton Community on Clouds and Climate via the Sulfur Cycle. Global Biogeochemical Cycles, 32(6), 1005–1026, doi: 10.1029/2017gb005862.
Wang, S.X. et al., 2014: Emission trends and mitigation options for air pollutants in East Asia. Atmospheric Chemistry and Physics, 14(13), 6571–6603, doi: 10.5194/acp-14-6571-2014.
Wang, X. et al., 2018: Field evidences for the positive effects of aerosols on tree growth. Global Change Biology, 24(10), 4983–4992, doi: 10.1111/gcb.14339.
Wang, Y. et al., 2019: Trends in particulate matter and its chemical compositions in China from 2013–2017. Science China Earth Sciences, 62(12), 1857–1871, doi: 10.1007/s11430-018-9373-1.
Wang, Z. et al., 2021: Incorrect Asian aerosols affecting the attribution and projection of regional climate change in CMIP6 models. npj Climate and Atmospheric Science, 4(1), 2, doi: 10.1038/s41612-020-00159-2.
Warner, J.X., Z. Wei, L.L. Strow, R.R. Dickerson, and J.B. Nowak, 2016: The global tropospheric ammonia distribution as seen in the 13-year AIRS measurement record. Atmospheric Chemistry and Physics, 16(8), 5467–5479, doi: 10.5194/acp-16-5467-2016.
Warner, J.X. et al., 2014: Global carbon monoxide products from combined AIRS, TES and MLS measurements on A-train satellites. Atmospheric Chemistry and Physics, 14(1), 103–114, doi: 10.5194/acp-14-103-2014.
Warner, J.X. et al., 2017: Increased atmospheric ammonia over the world’s major agricultural areas detected from space. Geophysical Research Letters, 44(6), 2875–2884, doi: 10.1002/2016gl072305.
Weber, J. et al., 2020: Minimal Climate Impacts From Short-Lived Climate Forcers Following Emission Reductions Related to the COVID-19 Pandemic. Geophysical Research Letters, 47(20), e2020GL090326, doi: 10.1029/2020gl090326.
Weber, R.J., H. Guo, A.G. Russell, and A. Nenes, 2016: High aerosol acidity despite declining atmospheric sulfate concentrations over the past 15 years. Nature Geoscience, 9(4), 282–285, doi: 10.1038/ngeo2665.
Weinstein, J.P., S.R. Hedges, and S. Kimbrough, 2010: Characterization and aerosol mass balance of PM2.5 and PM10 collected in Conakry, Guinea during the 2004 Harmattan period. Chemosphere, 78(8), 980–988, doi: 10.1016/j.chemosphere.2009.12.022.
Weisenstein, D.K., D.W. Keith, and J.A. Dykema, 2015: Solar geoengineering using solid aerosol in the stratosphere. Atmospheric Chemistry and Physics, 15(20), 11835–11859, doi: 10.5194/acp-15-11835-2015.
Wells, K.C. et al., 2020: Satellite isoprene retrievals constrain emissions and atmospheric oxidation. Nature, 585(7824), 225–233, doi: 10.1038/s41586-020-2664-3.
Wen, L. et al., 2018: Summertime fine particulate nitrate pollution in the North China Plain: increasing trends, formation mechanisms and implications for control policy. Atmospheric Chemistry and Physics, 18(15), 11261–11275, doi: 10.5194/acp-18-11261-2018.
Wennberg, P.O. et al., 2018: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products. Chemical Reviews, 118(7), 3337–3390, doi: 10.1021/acs.chemrev.7b00439.
West, J.J. et al., 2013: Co-benefits of mitigating global greenhouse gas emissions for future air quality and human health. Nature Climate Change, 3(10), 885–889, doi: 10.1038/nclimate2009.
Westervelt, D.M. et al., 2016: Quantifying PM2.5-meteorology sensitivities in a global climate model. Atmospheric Environment, 142, 43–56, doi: 10.1016/j.atmosenv.2016.07.040.
Westervelt, D.M. et al., 2018: Connecting regional aerosol emissions reductions to local and remote precipitation responses. Atmospheric Chemistry and Physics, 18(16), 12461–12475, doi: 10.5194/acp-18-12461-2018.
WHO, 2017: Evolution of WHO air quality guidelines: past, present and future. World Health Organization (WHO) Regional Office for Europe, Copenhagen, Denmark, 32 pp., www.euro.who.int/__data/assets/pdf_file/0019/331660/Evolution-air-quality.pdf .
Wichink Kruit, R.J. et al., 2017: Modelling trends in ammonia in the Netherlands over the period 1990–2014. Atmospheric Environment, 154, 20–30, doi: 10.1016/j.atmosenv.2017.01.031.
Wilcox, L.J. et al., 2019: Mechanisms for a remote response to Asian anthropogenic aerosol in boreal winter. Atmospheric Chemistry and Physics, 19(14), 9081–9095, doi: 10.5194/acp-19-9081-2019.
Wilcox, L.J. et al., 2020: Accelerated increases in global and Asian summer monsoon precipitation from future aerosol reductions. Atmospheric Chemistry and Physics, 20(20), 11955–11977, doi: 10.5194/acp-20- 11955-2020.
Wild, O. et al., 2020: Global sensitivity analysis of chemistry–climate model budgets of tropospheric ozone and OH: exploring model diversity. Atmospheric Chemistry and Physics, 20(7), 4047–4058, doi: 10.5194/acp-20-4047-2020.
Wilkinson, M.J. et al., 2009: Leaf isoprene emission rate as a function of atmospheric CO2 concentration. Global Change Biology, 15(5), 1189–1200, doi: 10.1111/j.1365-2486.2008.01803.x.
Williams, M.L. et al., 2018: The Lancet Countdown on health benefits from the UK Climate Change Act: a modelling study for Great Britain. The Lancet Planetary Health, 2(5), e202–e213, doi: 10.1016/s2542-5196(18)30067-6.
Wittig, V.E., E.A. Ainsworth, and S.P. Long, 2007: To what extent do current and projected increases in surface ozone affect photosynthesis and stomatal conductance of trees? A meta-analytic review of the last 3 decades of experiments. Plant, Cell & Environment, 30(9), 1150–1162, doi: 10.1111/j.1365-3040.2007.01717.x.
WMO, 2018: Scientific Assessment of Ozone Depletion: 2018. Global Ozone Research and Monitoring Project – Report No. 58, World Meteorological Organization (WMO), Geneva, Switzerland, 588 pp., https://csl.noaa.gov/assessments/ozone/2018/downloads/.
Woodward, S., D.L. Roberts, and R.A. Betts, 2005: A simulation of the effect of climate change-induced desertification on mineral dust aerosol. Geophysical Research Letters, 32(18), L18810, doi: 10.1029/2005gl023482.
Worden, H.M. et al., 2013: Decadal record of satellite carbon monoxide observations. Atmospheric Chemistry and Physics, 13(2), 837–850, doi: 10.5194/acp-13-837-2013.
Wu, S., L.J. Mickley, D.J. Jacob, D. Rind, and D.G. Streets, 2008: Effects of 2000–2050 changes in climate and emissions on global tropospheric ozone and the policy-relevant background surface ozone in the United States. Journal of Geophysical Research: Atmospheres, 113(D18), D18312, doi: 10.1029/2007jd009639.
Wu, X. et al., 2018: Characterization and source apportionment of carbonaceous PM2.5 particles in China – A review. Atmospheric Environment, 189, 187–212, doi: 10.1016/j.atmosenv.2018.06.025.
Xi, X. and I.N. Sokolik, 2016: Quantifying the anthropogenic dust emission from agricultural land use and desiccation of the Aral Sea in Central Asia. Journal of Geophysical Research: Atmospheres, 121(20), 12270–12281, doi: 10.1002/2016jd025556.
Xia, A.G., D. Michelangeli, and P.A. Makar, 2009: Mechanism reduction for the formation of secondary organic aerosol for integration into a 3-dimensional regional air quality model: α -Pinene oxidation system. Atmospheric Chemistry and Physics, 9(13), 4341–4362, doi: 10.5194/acp-9-4341-2009.
Xie, Y., M. Lin, and L.W. Horowitz, 2020: Summer PM2.5 pollution Extremes Caused by Wildfires Over the Western United States During 2017–2018. Geophysical Research Letters, 47(16), e2020GL089429, doi: 10.1029/2020gl089429.
Xie, Y. et al., 2018: Co-benefits of climate mitigation on air quality and human health in Asian countries. Environment International, 119, 309–318, doi: 10.1016/j.envint.2018.07.008.
Xu, L. and J.E. Penner, 2012: Global simulations of nitrate and ammonium aerosols and their radiative effects. Atmospheric Chemistry and Physics, 12(20), 9479–9504, doi: 10.5194/acp-12-9479-2012.
Xu, Y. and J.F. Lamarque, 2018: Isolating the Meteorological Impact of 21st Century GHG Warming on the Removal and Atmospheric Loading of Anthropogenic Fine Particulate Matter Pollution at Global Scale. Earth’s Future, 6(3), 428–440, doi: 10.1002/2017ef000684.
Xu, Y., D. Zaelke, G.J.M. Velders, and V. Ramanathan, 2013: The role of HFCs in mitigating 21st century climate change. Atmospheric Chemistry and Physics, 13(12), 6083–6089, doi: 10.5194/acp-13-6083-2013.
Yang, Y., S.J. Smith, H. Wang, S. Lou, and P.J. Rasch, 2019a: Impact of Anthropogenic Emission Injection Height Uncertainty on Global Sulfur Dioxide and Aerosol Distribution. Journal of Geophysical Research: Atmospheres, 124(8), 4812–4826, doi: 10.1029/2018jd030001.
Yang, Y., S.J. Smith, H. Wang, C.M. Mills, and P.J. Rasch, 2019b: Variability, timescales, and nonlinearity in climate responses to black carbon emissions. Atmospheric Chemistry and Physics, 19(4), 2405–2420, doi: 10.5194/acp-19-2405-2019.
Yang, Y. et al., 2016: Towards a quantitative understanding of total OH reactivity: A review. Atmospheric Environment, 134, 147–161, doi: 10.1016/j.atmosenv.2016.03.010.
Yang, Y. et al., 2020: Fast Climate Responses to Aerosol Emission Reductions During the COVID-19 Pandemic. Geophysical Research Letters, 47(19), e2020GL089788, doi: 10.1029/2020gl089788.
Yao, X. and L. Zhang, 2019: Causes of Large Increases in Atmospheric Ammonia in the Last Decade across North America. ACS Omega, 4(26), 22133–22142, doi: 10.1021/acsomega.9b03284.
Yarragunta, Y., S. Srivastava, and D. Mitra, 2017: Validation of lower tropospheric carbon monoxide inferred from MOZART model simulation over India. Atmospheric Research, 184, 35–47, doi: 10.1016/j.atmosres.2016.09.010.
Yienger, J.J. and H. Levy, 1995: Empirical model of global soil-biogenic NOx emissions. Journal of Geophysical Research: Atmospheres, 100(D6), 11447, doi: 10.1029/95jd00370.
Young, P.J. et al., 2013: Pre-industrial to end 21st century projections of tropospheric ozone from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmospheric Chemistry and Physics, 13(4), 2063–2090, doi: 10.5194/acp-13-2063-2013.
Young, P.J. et al., 2018: Tropospheric Ozone Assessment Report: Assessment of global-scale model performance for global and regional ozone distributions, variability, and trends. Elementa: Science of the Anthropocene, 6(1), 10, doi: 10.1525/elementa.265.
Yu, F., A.A. Nair, and G. Luo, 2018: Long-Term Trend of Gaseous Ammonia Over the United States: Modeling and Comparison With Observations. Journal of Geophysical Research: Atmospheres, 123(15), 8315–8325, doi: 10.1029/2018jd028412.
Yu, M. et al., 2019: Effects of air pollution control measures on air quality improvement in Guangzhou, China. Journal of Environmental Management, 244, 127–137, doi: 10.1016/j.jenvman.2019.05.046.
Yu, P., R. Xu, M.J. Abramson, S. Li, and Y. Guo, 2020: Bushfires in Australia: a serious health emergency under climate change. The Lancet Planetary Health, 4(1), e7–e8, doi: 10.1016/s2542-5196(19)30267-0.
Yu, P. et al., 2016: Radiative forcing from anthropogenic sulfur and organic emissions reaching the stratosphere. Geophysical Research Letters, 43(17), 9361–9367, doi: 10.1002/2016gl070153.
Yue, X. and N. Unger, 2014: Ozone vegetation damage effects on gross primary productivity in the United States. Atmospheric Chemistry and Physics, 14(17), 9137–9153, doi: 10.5194/acp-14-9137-2014.
Yue, X. et al., 2017: Ozone and haze pollution weakens net primary productivity in China. Atmospheric Chemistry and Physics, 17(9), 6073–6089, doi: 10.5194/acp-17-6073-2017.
Zakoura, M. and S.N. Pandis, 2018: Overprediction of aerosol nitrate by chemical transport models: The role of grid resolution. Atmospheric Environment, 187, 390–400, doi: 10.1016/j.atmosenv.2018.05.066.
Zanatta, M. et al., 2017: Source attribution of Arctic black carbon constrained by aircraft and surface measurements. Atmospheric Chemistry and Physics, 17(19), 11971–11989, doi: 10.5194/acp-17-11971-2017.
Zanis, P. et al., 2020: Fast responses on pre-industrial climate from present-day aerosols in a CMIP6 multi-model study. Atmospheric Chemistry and Physics, 20(14), 8381–8404, doi: 10.5194/acp-20-8381-2020.
Zare, A. et al., 2014: Quantifying the contributions of natural emissions to ozone and total fine PM concentrations in the Northern Hemisphere. Atmospheric Chemistry and Physics, 14(6), 2735–2756, doi: 10.5194/acp-14-2735-2014.
Zeng, G. et al., 2012: Trends and variations in CO, C2 h6, and HCN in the Southern Hemisphere point to the declining anthropogenic emissions of CO and C2 h6. Atmospheric Chemistry and Physics, 12(16), 7543–7555, doi: 10.5194/acp-12-7543-2012.
Zeng, G. et al., 2015: Multi-model simulation of CO and HCHO in the Southern Hemisphere: comparison with observations and impact of biogenic emissions. Atmospheric Chemistry and Physics, 15(13), 7217–7245, doi: 10.5194/acp-15-7217-2015.
Zhang, A. et al., 2020: Modeling the global radiative effect of brown carbon: A potentially larger heating source in the tropical free troposphere than black carbon. Atmospheric Chemistry and Physics, 20, 1901–1920, doi: 10.5194/acp-20-1901-2020.
Zhang, F. et al., 2012: Chemical compositions and extinction coefficients of PM2.5 in peri-urban of Xiamen, China, during June 2009–May 2010. Atmospheric Research, 106, 150–158, doi: 10.1016/j.atmosres.2011.12.005.
Zhang, G. et al., 2017: The single-particle mixing state and cloud scavenging of black carbon: a case study at a high-altitude mountain site in southern China. Atmospheric Chemistry and Physics, 17(24), 14975–14985, doi: 10.5194/acp-17-14975-2017.
Zhang, H., Y. Wang, T.-W. Park, and Y. Deng, 2017: Quantifying the relationship between extreme air pollution events and extreme weather events. Atmospheric Research, 188, 64–79, doi: 10.1016/j.atmosres.2016.11.010.
Zhang, H. et al., 2012: Source apportionment of PM2.5 nitrate and sulfate in China using a source-oriented chemical transport model. Atmospheric Environment, 62, 228–242, doi: 10.1016/j.atmosenv.2012.08.014.
Zhang, Q. et al., 2007: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophysical Research Letters, 34(13), L13801, doi: 10.1029/2007gl029979.
Zhang, Y. et al., 2016: Co-benefits of global and regional greenhouse gas mitigation for US air quality in 2050. Atmospheric Chemistry and Physics, 16(15), 9533–9548, doi: 10.5194/acp-16-9533-2016.
Zhang, Y. et al., 2019: Six-year source apportionment of submicron organic aerosols from near-continuous highly time-resolved measurements at SIRTA (Paris area, France). Atmospheric Chemistry and Physics, 19(23), 14755–14776, doi: 10.5194/acp-19-14755-2019.
Zhao, D. et al., 2019: Vertical characteristics of black carbon physical properties over Beijing region in warm and cold seasons. Atmospheric Environment, 213, 296–310, doi: 10.1016/j.atmosenv.2019.06.007.
Zhao, P.S. et al., 2013: Characteristics of concentrations and chemical compositions for PM2.5 in the region of Beijing, Tianjin, and Hebei, China. Atmospheric Chemistry and Physics, 13(9), 4631–4644, doi: 10.5194/acp-13-4631-2013.
Zhao, Y. et al., 2019: Inter-model comparison of global hydroxyl radical (OH) distributions and their impact on atmospheric methane over the 2000–2016 period. Atmospheric Chemistry and Physics, 19(21), 13701–13723, doi: 10.5194/acp-19-13701-2019.
Zhao, Y. et al., 2020a: Influences of hydroxyl radicals (OH) on top-down estimates of the global and regional methane budgets. Atmospheric Chemistry and Physics, 20(15), 9525–9546, doi: 10.5194/acp-20-9525-2020.
Zhao, Y. et al., 2020b: Substantial Changes in Nitrogen Dioxide and Ozone after Excluding Meteorological Impacts during the COVID-19 Outbreak in Mainland China. Environmental Science & Technology Letters, 7(6), 402–408, doi: 10.1021/acs.estlett.0c00304.
Zhao, Z. et al., 2019: Drought Impacts on Secondary Organic Aerosol: A Case Study in the Southeast United States. Environmental Science & Technology, 53(1), 242–250, doi: 10.1021/acs.est.8b04842.
Zheng, B. et al., 2018a: Rapid decline in carbon monoxide emissions and export from East Asia between years 2005 and 2016. Environmental Research Letters, 13(4), 44007, doi: 10.1088/1748-9326/aab2b3.
Zheng, B. et al., 2018b: Trends in China’s anthropogenic emissions since 2010 as the consequence of clean air actions. Atmospheric Chemistry and Physics, 18, 14095–14111, doi: 10.5194/acp-2018-374.
Zheng, B. et al., 2019: Global atmospheric carbon monoxide budget 2000-2017 inferred from multi-species atmospheric inversions. Earth System Science Data, 11(3), 1411–1436, doi: 10.5194/essd-11-1411-2019.
Zhou, Y., H. Mao, K. Demerjian, C. Hogrefe, and J. Liu, 2017: Regional and hemispheric influences on temporal variability in baseline carbon monoxide and ozone over the Northeast US. Atmospheric Environment, 164, 309–324, doi: 10.1016/j.atmosenv.2017.06.017.
Zhu, L. et al., 2015: Global evaluation of ammonia bidirectional exchange and livestock diurnal variation schemes. Atmospheric Chemistry and Physics, 15(22), 12823–12843, doi: 10.5194/acp-15-12823-2015.
Zhu, L. et al., 2017: Long-term (2005–2014) trends in formaldehyde (HCHO) columns across North America as seen by the OMI satellite instrument: Evidence of changing emissions of volatile organic compounds. Geophysical Research Letters, 44(13), 7079–7086, doi: 10.1002/2017gl073859.
Zou, Y., Y. Wang, Y. Zhang, and J.H. Koo, 2017: Arctic sea ice, Eurasia snow, and extreme winter haze in China. Science Advances, 3(3), e1602751, doi: 10.1126/sciadv.1602751.
Zusman, E. et al., 2013: Co-benefits: Taking a multidisciplinary approach. Carbon Management, 4(2), 135–137, doi: 10.4155/cmt.13.12.
1The word ‘concentration’ is used to denote abundances in terms of mixing ratio for most species unless specified.
2Prather et al. (2012) report lifetime due to stratospheric loss and soil uptake as 120 ± 24 years and 150 ± 50 years, respectively, and they were inadvertently swapped in AR5 with no effect on the total methane lifetime.
3The terms EC and BC are operationally defined on the basis of the methodology used for their quantification (i.e., thermal refractivity and light absorption, respectively) and often used interchangeably.
4The carbon mass fraction of OA is termed as OC, a conversion factor between 1.4 to 1.6 from OC to OA is typically assumed.
5Baseline ozone is defined as the observed ozone at a site when it is not influenced by recent, locally emitted or anthropogenically produced pollution (Jaffe et al., 2018).
Table of Contents





